MDCG 2019-16 rev.1:医疗器械网络安全指南
文件编号:MDCG 2019-16 rev.1
发布日期:2020年12月(rev.1)
适用法规:EU MDR 2017/745 / EU IVDR 2017/746
官方PDF:下载
文件目的
本指南为医疗器械(包括医疗器械软件MDSW)的网络安全要求提供实践指导,帮助制造商满足EU MDR/IVDR中关于网络安全的GSPR要求。
网络安全的法规基础
GSPR相关要求
EU MDR附件I(GSPR)中与网络安全相关的要求:
- 第17.2节:含有电子可编程系统的器械须设计为确保可重复性、可靠性和性能
- 第17.4节:须保护免受未经授权访问
- 网络安全须纳入风险管理(EN ISO 14971)
适用范围
本指南适用于:
- 医疗器械软件(MDSW)
- 含有软件的医疗器械
- 连接网络的医疗器械(包括IoMT)
网络安全生命周期
安全开发生命周期(SDLC)
制造商须将网络安全整合到产品开发全生命周期:
设计阶段:
- 安全架构设计
- 威胁建模(STRIDE方法)
- 安全需求定义
开发阶段:
- 安全编码规范
- 代码审查
- 静态代码分析
验证阶段:
- 渗透测试
- 漏洞扫描
- 安全功能测试
上市后阶段:
- 漏洞监测
- 安全补丁管理
- 事件响应计划
技术文件要求
须包含的网络安全文件
| 文件类型 | 内容要求 |
|---|---|
| 安全架构文档 | 系统架构、数据流图、信任边界 |
| 威胁建模报告 | 识别的威胁、攻击面分析 |
| 风险评估 | 网络安全风险与安全措施 |
| 安全测试报告 | 渗透测试、漏洞扫描结果 |
| 上市后网络安全计划 | 漏洞监测、补丁管理流程 |
最低安全要求
认证与授权:
- 强密码策略
- 多因素认证(高风险器械)
- 基于角色的访问控制
数据保护:
- 传输加密(TLS 1.2+)
- 存储加密(敏感数据)
- 数据完整性验证
审计日志:
- 记录安全相关事件
- 日志防篡改保护
- 日志保留策略
软件更新:
- 安全的软件更新机制
- 更新完整性验证
- 回滚能力
与AI法案(AIA)的关系
参见 MDCG 2025-6: MDR/IVDR与AI法案互动FAQ
相关文件
官方文件全文
MDCG 2019-16 Rev.1 Guidance on Cybersecurity for medical devices
December 2019 July 2020 rev.1
This document has been endorsed by the Medical Device Coordination Group (MDCG) established by Article 103 of Regulation (EU) 2017/745. The MDCG is composed of representatives of all Member States and it is chaired by a representative of the European Commission.The document is not a European Commission document and it cannot be regarded as reflecting the official position of the European Commission. Any views expressed in this document are not legally binding and only the Court of Justice of the European Union can give binding interpretations of Union law.
1. Introduction
1.1. Background
The two new Regulations on medical devices 745/2017 (MDR) and 746/2017 (IVDR) (hereafter called the Medical Devices Regulations) have been adopted and entered into force on 25 May 2017. The two Regulations, which are to replace three EU Directives 1 , apply progressively until May 2021 for medical devices and May 2022 for in vitro diagnostic medical devices.
Among the many novelties introduced, the two Regulations enhance the focus of legislators on ensuring that devices placed on the EU market are fit for the new technological challenges linked to cybersecurity risks. In this respect, the new texts lay down certain new essential safety requirements for all medical devices that incorporate electronic programmable systems and software that are medical devices in themselves. They require manufacturers to develop and manufacture their products in accordance with the state of the art taking into account the principles of risk management, including information security, as well as to set out minimum requirements concerning IT security measures, including protection against unauthorised access.
1.2. Objectives
The primary purpose of this document is to provide manufacturers with guidance on how to fulfil all the relevant essential requirements of Annex I to the MDR and IVDR with regard to cybersecurity. However, and in light of the complexity of medical device supply chains and the role played by different operators in ensuring that devices are protected against unauthorised access and possible cyber threats, additional considerations concerning expectations from actors other than manufacturers are provided. In addition, a description of other EU and global pieces of legislation and guidance that are relevant to the domain of cybersecurity for medical devices has been provided in an Annex.
1.3. Cybersecurity Requirements included in Annex I of the Medical Devices Regulations
Cybersecurity requirements listed in Annex I of the Medical Devices Regulations, deal with both premarket and post-market aspects. These requirements, and their interconnection, are illustrated in Figure 1 and are elaborated in Chapter 2 with the aim to provide a basis for the development of recommendations and guidance for medical device manufacturers (Chapters 3-6 of this document).
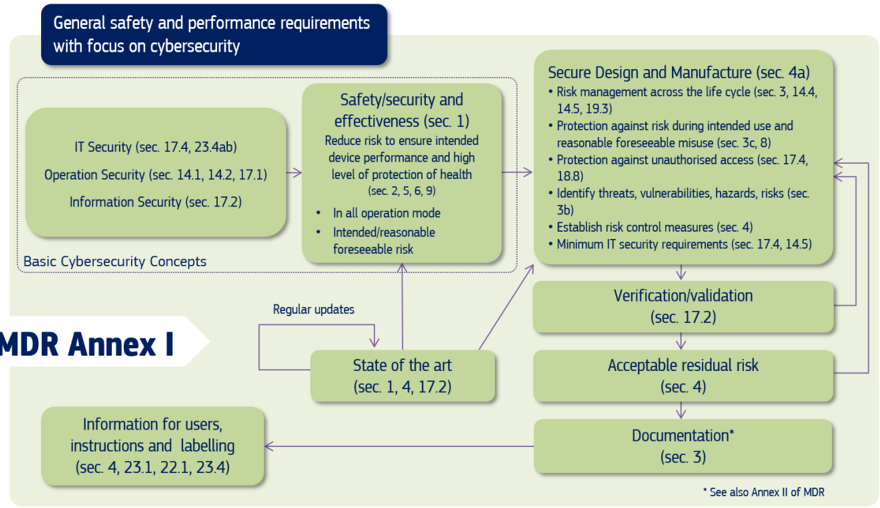
Figure 1: Cybersecurity requirements contained in MDR Annex I
The above requirements illustrated in Figure 1, are also applicable to those included in Annex I of Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR). The correspondence between the sections in MDR Annex I and IVDR Annex I relevant for this guidance is provided in Table 1.
Table 1: Correspondence table between sections, relevant for this guidance, in MDR Annex I and IVDR Annex I
| Main topic | Section number MDRAnnex I | Section number IVDR Annex I |
|---|---|---|
| Device performance | 1 | 1 |
| Risk reduction | 2 | 2 |
| Risk management system | 3 | 3 |
| Risk control measures | 4 | 4 |
| Minimisation of foreseeable risks, and any undesirable side-effects | 8 | 8 |
| Combination/connection of devices/systems | 14.1 | 13.1 |
| Interaction between software and the IT environment | 14.2.d | 13.2.d |
| Interoperability and compatibility with other devices or products | 14.5 | 13.5 |
| Repeatability, reliability and performance | 17.1 | 16.1 |
| Development and manufacture in accordance with the state of the art taking into account the principles of development life cycle, risk management, including information security, verification and validation | 17.2 | 16.2 |
| Minimum IT requirements | 17.4 | 16.4 |
| Unauthorised access | 18.8 | - |
| Lay persons | 22.1 | - |
| Residual risks (information supplied by the manufacturer) | 23.1 g | 20.1 g |
| Warnings or precautions (information on the label) | 23.2 m | 20.2 m |
| Residual risks, contra-indications and any undesirable side-effects, (information in the instructions for use) | 23.4 g | - |
| Minimum IT requirements (information in the instructions for use) | 23.4.ab | 20.4.1.ah |
MDR
Cybersecurity Act
1.4. Other Cybersecurity Requirements
Several requirements that are generally associated with cybersecurity are not explicitly mentioned in the Medical Devices Regulations. Of particular relevance are those requirements regarding privacy and confidentiality of data associated with the use of MDs that may be outside the scope of the Medical Devices Regulations but are subject to other legislations (see Chapter 7).
In the context of cybersecurity and within the MDR, the manufacturer should be particularly aware of the following provisions (also illustrated in Figure 2): Documentation
- Privacy and data protection: Article 62.4(h): General requirements regarding clinical investigations conducted to demonstrate conformity of devices
- Conformity assessment procedures : Article 52
- Post-market surveillance system of the manufacturer: Article 83
- Post-market surveillance plan: Article 84
- Post-market surveillance report: Article 85
- Periodic safety update report: Article 86
- Reporting of serious incidents and field safety corrective actions: Article 87
- Trend reporting: Article 88
- Analysis of serious incidents and field safety corrective actions: Article 89
- Technical documentation: Annex II
- Technical documentation on post-market surveillance: Annex III
- Clinical evaluation and post-market follow-up: MDR Chapter VI and Annex XIV
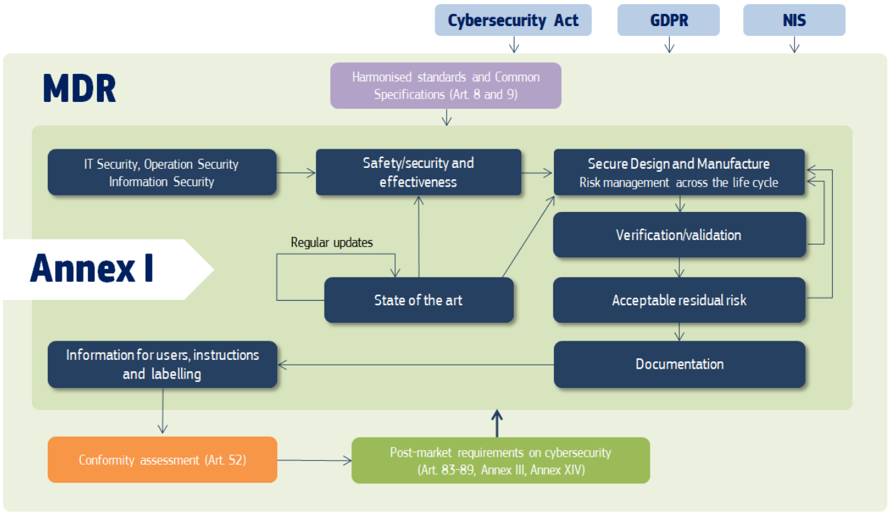
Figure 2: Cybersecurity requirements in the MDR; the application of other relevant EU legislations, such as Cybersecurity Act, GDPR and NIS is discussed in more detail in Chapter 7
GDPR
NIS
As shown in Figures 1 and 2, the Medical Devices Regulations request manufacturers of medical devices to consider the state of the art when designing, developing and upgrading medical devices across their life cycle. Manufacturers should demonstrate state-of-the-art within their decisions (based on applicable standards, guidance, their own proprietary knowledge and publicly available scientific / technical information) while demonstrating appropriateness to proportionally address security risk.
Table 2 provides an overview of the particular focus that should be placed on managing cybersecurity across the entire life cycle of a medical device. Additionally, the table demonstrates the different activities that the manufacturer needs to carry out.
Table 2: Cybersecurity activities across the life cycle of medical devices according to the Medical Devices Regulations
| Pre-market activities | Post-market activities |
|---|---|
| Secure Design (Annex I) | |
| Risk management (Annex I) | Risk management (Annex I) |
| Establish Risk Control Measures (Annex I) | Modify Risk Control Measures /Corrective Actions/Patches (Annex I) |
| Validation, Verification, Risk Assessment, Benefit Risk Analysis (Annex I) | Validation, Verification, Risk Assessment, Benefit Risk Analysis (Annex I) |
| Technical Documentation (Annex II and III) | Maintain and update a Post-market Surveillance Plan and Post-market Surveillance System (Article 83 and 84) |
| Conformity Assessment (Article 52) | Trend Reporting (Article 88) |
| Establish a Post-market Surveillance Plan and Post-market Surveillance System (Article 83 and 84) | Analysis of Serious Incidents (Article 89) |
| Clinical evaluation process (Chapter VI) | Post-Market Surveillance Report (Article 85) |
| Periodic Safety Update Report (Article 86) | |
| Update Technical Documentation (Annex II and III) | |
| Inform the Electronic System On Vigilance (Article 92) |
1.5. Abbreviations
| Abbreviation | Meaning |
|---|---|
| CE | Clinical Evaluation |
| CIA | Confidentiality, Integrity and Availability |
| CSIRT | Computer Security Incident Response Team |
| EN | European Standard |
| ENISA | European Union Agency for Cybersecurity |
| FSCA | Field Safety Corrective actions |
| GDPR | General Data Protection Regulation |
| IEC/TR | International Electrotechnical Commission - Technical Report |
| GSPR | General Safety and Performance Requirements |
| IMDRF | International Medical Device Regulators Forum |
| ISMS | Information security management system |
| ISO/IEC | International Organisation for Standardisation/ International Electrotechnical Commission |
|---|---|
| IT | Information Technology |
| IVDR | In Vitro Diagnostic Medical Devices Regulation; EU 2017/746 |
| MD | Medical Device |
| MDCG | Medical Device Coordination Group |
| MDR | Medical Devices Regulation; EU 2017/745 |
| MDS2 | Manufactures Disclosure Statement for Medical Device Security |
| MDSW | Medical Device Software |
| MIR | Manufacturer Incident Report |
| NIS | Network and Information Security |
| NIST | National Institute of Standards and Technology |
| OES | Operator of Essential Services |
| OTS | Off the Shelf software |
| PSR | Periodic Summary Reports |
| PSUR | Periodic Safety Update Report |
| QMS | Quality Management System |
| SOTA | State of the Art |
2. Basic Cybersecurity Concepts
A central definition relevant to all cybersecurity requirements within the Medical Devices Regulations is that of "risk" 2 :
'risk' means the combination of the probability of occurrence of harm and the severity of that harm
Such a definition is all encompassing by nature and applies to several types of risks. This is intentionally done so that to fulfil the primary protection goals laid out in the Regulations. It is acknowledged that in the field of medical devices, (security) risk has to be reduced to an acceptable level.
2.1. IT Security, Information Security, Operation Security
Annex I of the Medical Devices Regulations explicitly sets out the requirement for manufacturers of in vitro diagnostic medical device and medical device to fulfil minimum requirements concerning hardware, IT networks 3 characteristics and IT security measures, including protection against unauthorised access. All these requirements are necessary in order to run the software as intended (see sections 17.4, 18.8 and 23.4b in MDR and 16.4 and 20.4.1(c) in the IVDR).
IT Security: this term is generally understood as the protection of computer systems from adverse effects on assets including hardware, software or electronic data, as well as from disruption or misdirection of the services they provide. In relation to IT security, ENISA 4 defines the term Communication Security Domain as "Protection against a threat to the technical infrastructure of a cyber system which may lead to an alteration of its characteristics in order to carry out activities which were not intended by its owners, designers or users".
Key concepts involved in IT security specifically for medical devices are the following:
Confidentiality of information at rest and in transit
Integrity, which is necessary to ensure information authenticity and accuracy (i.e. nonrepudiation)
Availability of the processes, devices, data, and connected systems
It is important to ensure confidentiality dependent on the risk management, e.g. if the state of the healthcare situation or condition is critical in conjunction with the significance of information provided by the product function.
Operation Security: ENISA 5 defines Operation Security Domain as "Protection against the intended corruption of procedures or workflows which will have results that were unintended by its owners, designers or users". The MDR Annex I sections 17.4, 18.8, and IVDR Annex I section 16.4 address issues related to the operating security of the IT infrastructure of medical devices.
Information Security: Annex I section 17.2 (MDR) or 16.2 (IVDR) explicitly sets out that devices which incorporate software or software that are devices in themselves, the software shall be developed and manufactured in accordance with the state of the art taking into account the principles of development life cycle, risk management, including information security, verification and validation. ENISA 6 defines Information Security Domain as "Protection against the threat of theft, deletion or alteration of stored or transmitted data within a cyber system". Other available definitions involve further issues and concepts, such as Confidentiality, Integrity and Availability (CIA).
2.2. Safety, Security and Effectiveness
Information security and IT security are addressed explicitly in Annex I 17.2 (MDR), 17.4 (MDR), 18.8 (MDR), 16.2 (IVDR) and 16.4 (IVDR) whereas "Safety and Effectiveness" issues are addressed in section 1 of Medical Devices Regulations Annex I:
Devices shall achieve the performance intended by their manufacturer and shall be designed and manufactured in such a way that, during normal conditions of use, they are suitable for their intended purpose. They shall be safe and effective and shall not compromise the clinical condition or the safety of patients, or the safety and health of users or, where applicable, other persons, provided that any risks which may be associated with their use constitute acceptable risks when weighed against the benefits to the patient and are compatible with a high level of protection of health and safety, taking into account the generally acknowledged state of the art.
Overall, Annex I section 1 of the Medical Devices Regulations requests that any risks associated with the operation of medical devices must be acceptable so as to enable a high level of protection of health and safety. This can be only achieved through the establishment of an adequate balance between benefit and risk during all possible operation modes of a medical device. To this end, there is a need to consider the relationship between "safety and security" as they relate to risk. As illustrated below in Figure 3 patients' safety may be compromised due to "security issues" which may have "safety impacts".
breach of data and systems security and reduction of effectiveness)
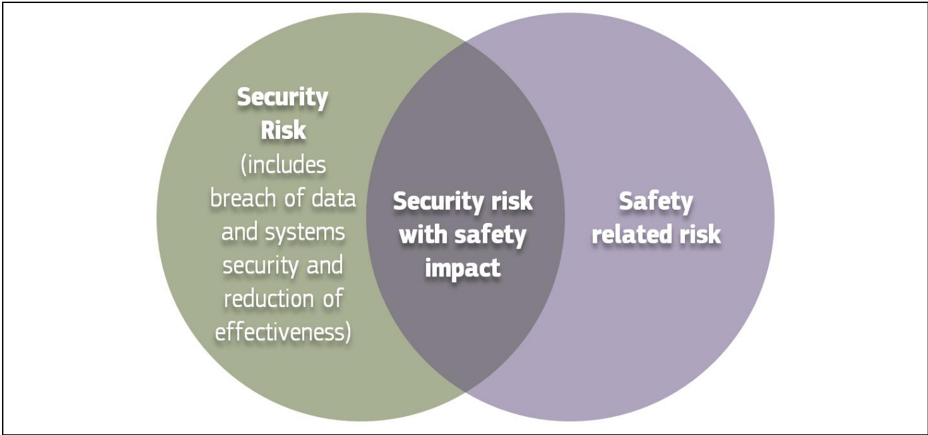
Figure 3: Cybersecurity measures may cause safety impacts
Security issues may be of both weak and/or restrictive security:
- a) Weak security: for example, weak access control may allow malicious modification of the operation of an implanted cardiac device.
- b) Restrictive security: the use of too restrictive security measures that provide a high level of protection may have a safety impact, especially if the security functionalities are not well designed. For example, during an emergency, the medical personnel must be able to access an implanted cardiac device without restrictions, but strong security measures need to be in place under normal operating conditions.
Therefore, when assessing risks in accordance with Annex I of the Medical Devices Regulations, it is important to include security issues in the risk assessment, even in cases where security is not stated explicitly in the Regulations' requirements (for example Annex I: 1, 2, 3d, 4a, 8, 9, 17.1, 18.1 (MDR) or 14.1 (IVDR)) on risk minimisation (those risks may also include security risks with safety impacts).
2.3. Intended use and intended operational environment of use
Manufacturers determine design inputs associated with cybersecurity requirements to ensure safety and effectiveness of products against cybersecurity risks and threats. These cybersecurity requirements should be considered in accordance with the nature of the device, including the device type and intended communication technologies usage. In specific circumstances, and depending on the device type, intended use and intended operational environment, the manufacturer may decide (based on a safety and security risk assessment) to implement less strict security controls.
A medical device should be designed in a layered defence in depth approach and therefore should not rely on security controls in the operating environment. Nevertheless, as part of this layered defence in depth approach, there are expectations on the intended operating environment (see also the Chapter 3 of this guidance). Expectations on the operating environment might include protection and performance characteristics. Often the expectations are common best practice, often called 'good security hygiene'. Expectations on the intended operating environment should be clearly documented and communicated to the operator.
2.4. Reasonably foreseeable misuse 7
Due to the complexity of software development, software configuration and interdependencies between software components, cybersecurity vulnerabilities exist in most products. Whether or not a vulnerability will be discovered and if it will be exploited is unknown until it occurs. The general assumption is that any vulnerability which is deemed to be exploitable for a given implementation of software, might be discovered and exploited over time and as such should be regarded as an enabler for reasonably foreseeable misuse.
Medical device manufacturers should ensure that a medical device is designed and manufactured in a way that ensures that the risks associated with reasonably foreseeable environmental conditions are removed or minimised 8 . This may include the infield monitoring of the software's vulnerabilities and the possibility to perform a device update (outside the context of a field safety corrective action) through, for example delivering patches to ensure the continued security of the device.
During the risk management process, the manufacturer should foresee or evaluate the potential exploitation of those vulnerabilities that may be a result of reasonably foreseeable misuse. This, however, may depend on the specific situation. For example, using an unsecured memory-stick to enter data into a medical IT system can be considered 'reasonably foreseeable misuse', while the input of x-ray images via a CD may be considered 'intended use'. Due to the huge variety of use environments, this decision may even depend on the specific installation and use environment.
During the product security risk management process, the manufacturers need to distinguish two important areas:
- Safety risk management normally covered in the overall product risk management, and
- Security risk, which is not associated to safety.
2.5. Operating Environment
Devices operating in the intended use environment should consider that the IT infrastructure of the different healthcare providers has unique and different risk management approaches associated with their networks. Healthcare providers should adopt a risk management process adhering to general cybersecurity best practices to maintain the healthcare provider's overall security status, among others, as follows:
Good physical security to prevent unauthorised physical access to medical device or network access points;
Access control measures (e.g. role based) to ensure only authenticated and authorised personnel are allowed access to network elements, stored information, services and applications;
Network access controls, such as segmentation, to limit medical device communication;
General patch management practices that ensure timely security patch updates;
Malware protection to prevent unauthorised code execution;
Security awareness training.
Auditability that supports non-repudiation, i.e. the ability to reliability determine who made what changes to the system and when to assist with forensics
2.6. Joint Responsibility - Specific expectations from other stakeholders
While the MDR and the IVDR provide legal obligations only with regard to manufacturers, however it should be noted that for the provision of secured healthcare services, it is important to recognise the roles and expectations of all stakeholders, such as manufacturers, suppliers, healthcare providers, patients, integrators, operators and regulators. All of these actors share responsibilities for ensuring a secured environment for the benefit of patients' safety. For example, agreements contemplating responsibility are one option to ensure that all parties understand the joint responsibility of managing devices in a medical IT-network.
Specific expectations from stakeholders in the field of cybersecurity are also analysed and listed by the IMDRF document IMDRF/CYBER WG/N 60, which is still at its drafting stage. This guidance is in substantial alignment with those IMDRF preliminary considerations 9 10 .
Modification of a medical device, e.g. the installation or enabling of third-party software including software patching, should always be under explicit published guidance of the manufacturer. It is important to understand that any invalidated modification of a medical device or system (e.g., product firewall changes, software patches, security software, utilities, games, music files, other software programs, etc.) can adversely affect system performance or safety in unpredictable ways. For example, it may open doors for easy exploitation of identified vulnerabilities of the medical device.
2.6.1. Integrator
Integrating a medical device may often enable practical features of other network components towards a more efficient use of the existing functions of a connectable medical device. Integration has the potential to improve information security because it will allow the implementation of additional technical protection measures that have to be based on the specific integration environment, e.g. authenticated communication nodes and authenticated users and roles, and encrypted data flow. The integrator is contracted either by the manufacturer or the operator. All legal responsibilities (e.g. in the domain of Product Safety, GDPR, MDR or IVDR) for the safe functioning of the integrated system remain with the entity that has a contract with the integrator, to the extent permitted by existing EU or national law.
In the case where a medical device manufacturer contracts out the integration of a device on the user's site, then all obligations and liabilities resulting from that integration remain with the manufacturer. This does not exempt the customer from their responsibilities of compliance with any other regulations applicable to them (e.g. the national transposition of the NIS Directive in the case of OES).
Consistently, obligations towards establishing information security of a specific integrated device effectively remain with the Health Delivery Organisation if it has mandated the integrator to connect a given medical device to the clinical/hospital IT network.
The main responsibility of the integrator is the installation and configuration of the system and the integration into the operator's environment. The integrator should ensure that the system is configured in such a way that it can operate securely in the health and medical service target environment. Integration per se does not alter or extend the intended use of the essential medical functions of the individual medical devices.
- Assess reasonable level of security for the operating environment;
- Integrate the system into the environment at the operator site, including secure configuration of the system;
- Provide required documentation and training to operator and operator personnel;
- Provide support for patching and security incident handling.
2.6.2. Operator
Devices should be used as intended by the manufacturer, following the instructions for use provided with the devices. The operator should follow the manufacturers' published requirements and guidelines regarding security for commissioning, operating and de-commissioning of medical devices, e.g. isolate a medical device from the internet if not required for its operation; apply software patches per the manufacturer's instruction (when this is the responsibility of the operator) or ensure that antimalware software is up-to-date, if applicable.
The operator needs to contact the manufacturer if an appropriate set of security information is not available, e.g. security information in the Instructions for Use or provided in separate documents such as the Manufacturers Disclosure Statement for Medical Device Security (MDS2), installation guides or any other form of documentation.
The operator is responsible for the procurement and should ensure that security is maintained during the operation and application of the system (medical device), and particularly not compromised by changes in the environment of by user interaction.
- Ensure required level of security for operational environment (network, physical, …);
- Provide required infrastructure (network, physical);
- Ensure that personnel are properly trained and available in case of security issues;
- Ensure that system is used as proscribed by manufacturer guidelines (e.g. no physical access by unauthorized users, password policies kept, network security measures);
- Ensure that prescribed maintenance is done as required, including installation of security patches;
- Notify the manufacturer without delay of any suspected security event.
2.6.3. Users including healthcare & medical professionals, patients & consumers
Healthcare and medical professionals (including medical doctors, nurses, radiologists and radiographers, pathologists, etc.) are responsible for the use of medical devices for their purposes, e.g. to diagnose, prevent, monitor, treat or alleviate disease or injury patients. These users may access, review and exchange data with the devices, and may be responsible for the patient's education and establishing software and devices parameters of usage.
Patients and consumers are encouraged to employ cyber smart behaviour, such as paying attention to privacy, being aware of suspicious messaging, and browsing responsibly. Instruction for Use should include the necessary information so that patients and consumers can be up-to-date with the latest ess able a bug out the chine me cycle.
Design and Development version of software, protect the device throughout its lifespan, use sufficiently complex passwords, turn off features that are not used, secure the computer or tablet devices, use backups and protection of their healthcare data. This includes ensuring that connected devices, such as computers and mobile devices comply with the operating instructions provided with the medical device. These provisions will ensure secure coexistence of medical devices in an Internet of Things (IoT) or an Internet of Devices (IoD) environment. Instructions for use should take into account the age or other limiting factors of the intended users. Methods for authentication and authorisation should be appropriate to the device.
3. Secure Design and Manufacture
Safety, security and effectiveness are critical aspects in the design of security mechanisms for in vitro diagnostic medical devices and medical devices. Therefore, there is a clear requirement that these aspects need to be considered by the manufacturers from an early stage of development and manufacturing process and throughout the entire life cycle. 11
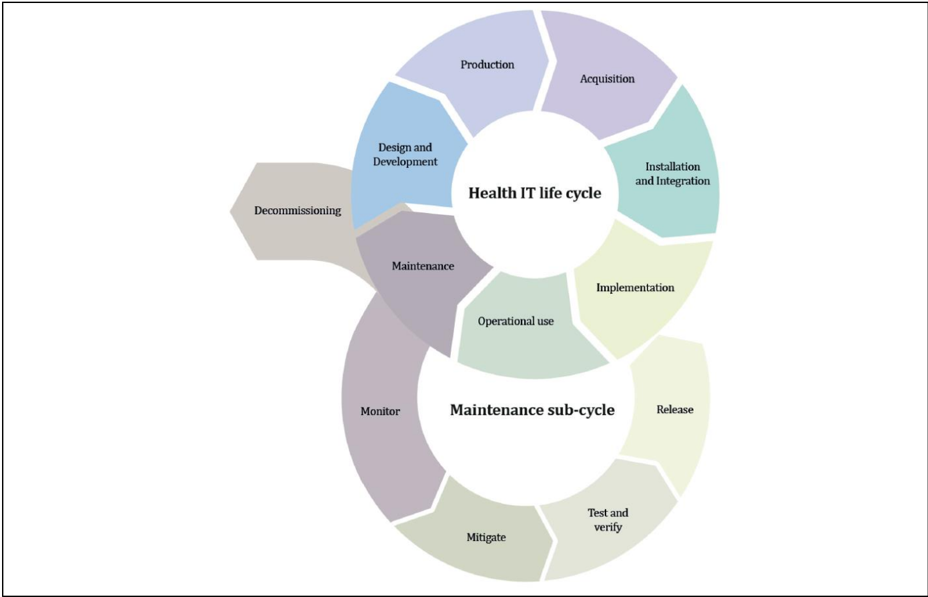
Figure 4 : Life cycle stages
- Annex I Section 4 of IVDR/MDR: adopt risk control measures for the design and manufacture of the devices conforming to safety principles and taking account of the generally acknowledged state of the art.
Annex I Section 16 (IVDR) or 17 (MDR) on "Electronic programmable systems - devices that incorporate electronic programmable systems and software that are devices in themselves"
Annex I Sections 16.4 (IVDR) or 17.4 and 18.8 (MDR) making explicit reference on the issues of "minimum requirements concerning hardware, IT networks characteristics and IT security measures", including protection against unauthorised access " protection from unauthorised access " as a key cybersecurity control measure
Annex I Sections 22.1 (MDR) making explicit reference on the issues of "devices for use by lay person' where it is stated that this type of medical devices 'shall be designed and manufactured in such a way that they perform appropriately for their intended purpose taking into account the skills and the means available to lay persons and the influence resulting from variation that can be reasonably anticipated in the lay person's technique and environment "
3.1. 'Secure by design'
Figure 5 illustrates how 'secure by design' practices in this document contribute to a 'defence in depth' strategy for the product. The 'security management' practice is shown on the top circle since it is applied throughout all other practices to ensure that these practices are being followed and managed. The other practices, shown on the bottom circle are applied throughout the development lifecycle, often in an iterative pattern. These practices each contribute to the overall 'defence in depth' strategy which is shown as the centre of the circle because it represents the key result of following the security development lifecycle.
Defect management and security update management provide verified repairs to secure implementation and fall under the category of overall security management in the diagram.
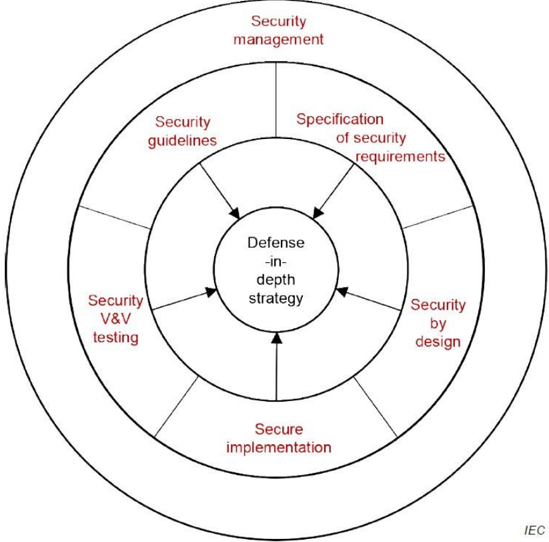
Figure 5: Defence in depth strategy is a key philosophy of the secure product life-cycle
The overall approach for security management for medical devices and software does not differ from the security management of other cyber-physical systems. The following common eight practices define most of the necessary processes that should be in place to establish a 'Defense-in-Depth strategy' for the organisation during the product lifecycle:
Practice 1 - Security management: The purpose of the security management practice is to ensure that the security-related activities are adequately planned, documented and executed throughout the product's lifecycle.
Practice 2 - Specification of security requirements: The processes specified by this practice are used to identify the security capabilities that are required for appropriate protection of confidentiality, integrity and availability of data, function and services of the medical device along with the specified product security context. Security capabilities can include such items as authentication, authorisation, encryption, auditing and other security capabilities a product needs to include. The product security context can include items such as physical security level, protection of external interfaces via a
Medical Device Coordination Group Document firewall, etc. These security requirements can be defined at the product-level or they may supplement product-level requirements.
Practice 3 - Secure by design: The processes specified by this practice are used to ensure that the product is secure by design including defence in depth.
Practice 4 - Secure implementation: The processes specified by this practice are used to ensure that the product features are implemented securely. Requirements in this practice apply to all hardware and software components in the product with the exception of externally provided components. For externally provided components, requirements of Practice 1 (Security Management) apply instead.
Practice 5 - Security verification and validation testing: The processes specified by this practice are used to document the security testing required to ensure that all the security requirements have been met for the product and that security of the product is maintained when the product is used as intended. Security testing should be aligned to other product test activities, and can be performed at various times by various personnel during the total security lifecycle based on the type of testing and the development model used by the vendor.
Practice 6 - Management of security-related issues: The processes specified by this practice are used for handling security-related issues of a product.
Practice 7 - Security update management: The processes specified by this practice are used to ensure that security updates and security patches associated with the product are tested for regressions and made available to product users in a timely manner.
Practice 8 - Security guidelines: The processes specified by this practice are used to provide and maintain user documentation that describes how to integrate, configure, and maintain the defence in depth strategy of the product in accordance with its product security context.
3.2. Security Risk Management
Risk Management is generally understood as the discipline of identifying and measuring risks towards safety and effectiveness resulting from the intended use and foreseeable misuse of a medical device and reducing them "as far as possible" to an acceptable level (see Annex I, sections 16.1 (IVDR) or 17.1 (MDR)). The general approach to risk management for medical devices according to the state-ofthe-art can be found in the Medical Devices Regulations Annex I, Section 3 and relevant harmonised standards published in the Official Journal.
Risks related to data and systems security are specifically mentioned within the scope of the risk management process, to avoid any misunderstanding that a separate process would be needed to manage security risks related to medical devices. Specific methods and requirements are however used for security risks.
The security risk management process has the same elements as safety risk management process, all documented in a security risk management plan. The process elements are security risk analysis, security risk evaluation, security risk control, evaluation of residual security risk and reporting. When a security risk or control measure could have a possible impact on safety and effectiveness, then it should be included in the safety risk assessment. Similarly, any safety risk control or consideration that might have an impact on security should be included in the security risk analysis (see Annex IV for a descriptive illustration of this concept)
Describe intended use,
(sec. 4)
Reduce risks:
As an example, 'blanking' a screen might be an appropriate security control to mitigate the disclosure of personal data, but when the medical device is used for interventional use or the display of vital signs, then 'blanking' the screen is a safety concern and thus should not be implemented.
Chapter 2.2 of this guidance describes how security vulnerabilities may affect the product's safety or effectiveness. A product risk analysis for safety should therefore consider the effects of security vulnerabilities to the essential functioning of the product. The safety risk assessment might list generic security related hazards identified for the product, such as but not limited to: denial of service, execute code, memory corruption, gain information, gain privilege, etc. This is to avoid detailing every possible security attack vector which does not result in a different hazard for the product.
The risks to be addressed in Annex I, sections 3 and 4 of the Medical Devices Regulations, refer to both safety and security issues and the overall information flow can be illustrated in the below Figure 6. Review before product release: Evaluate all risks together
• Review information on
- Art 83-89, Annex III, XIV
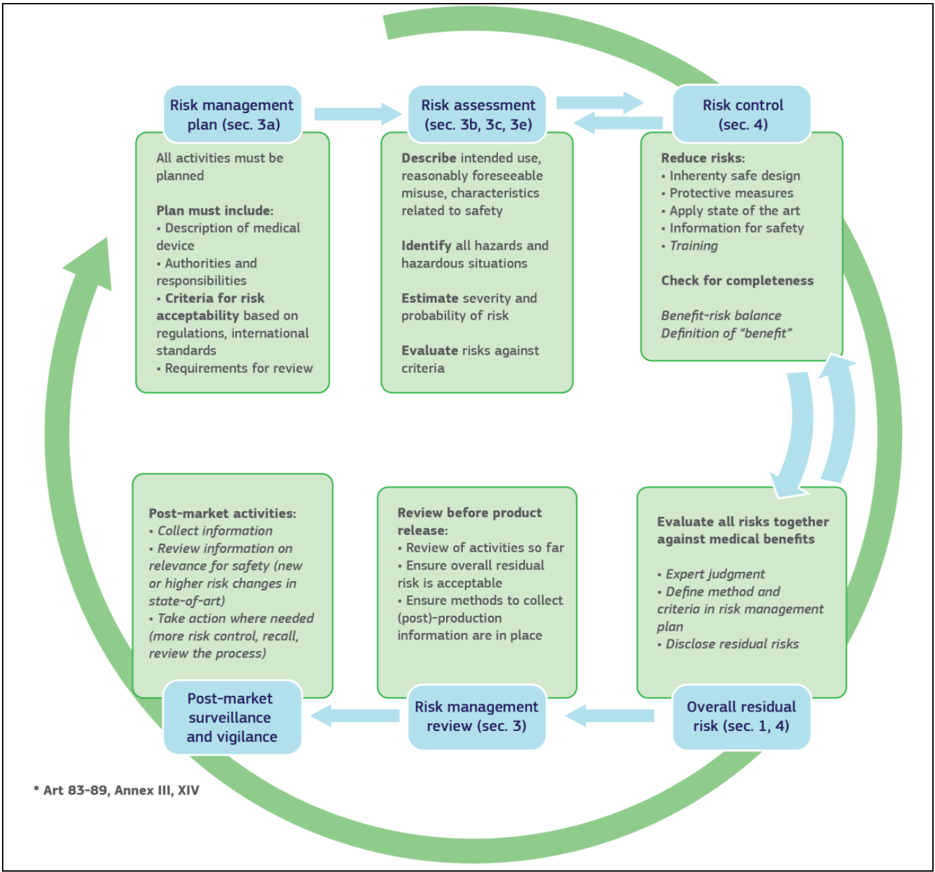
Figure 6: Information flow in safety and security risk management for MDs
• Review of activities so far against medical benefits
3.3. Security Capabilities
The list of known vulnerabilities and attack vectors is the basis for specifying the security capabilities, depending on the risk management, required for appropriate protection of confidentiality, integrity, availability of data, function and services of the medical device along with the specified product security context.
Security capabilities may be determined as suitable risk-control measures. The design and implementation of such capabilities need to comply with the state of the art (see Annex I, sections 17.2 (MDR) or 1, 4, 16.2 (IVDR)) and cover a wide range of technical areas (see Table 3).
An indicative list of security capabilities which can be used to protect the device and establish a means for appropriate communication with the operator is provided in Table 3.
Table 3: Indicative list of security Capabilities for MD
| Automatic Logoff |
|---|
| Audit Controls |
| Authorization |
| Configuration of Security Features |
| Cybersecurity Product Upgrades |
| Personal Data De-Identification |
| Data Backup and Disaster Recovery |
| Emergency Access |
| Personal Data Integrity and Authenticity |
| Malware Detection / Protection |
| Node Authentication |
| Person Authentication |
| Physical Locks |
| System and OS Hardening |
| Security and Privacy Guides |
| Personal Data Storage Confidentiality |
| Transmission Confidentiality |
| Transmission Integrity |
Where there is an impact on safety or effectiveness 12 , manufacturers shall select the most appropriate risk control solution, in the following order of priority:
- a) Eliminate or reduce risks as far as possible through safe design and manufacture;
- b) Where appropriate, take adequate protection measures, including alarms if necessary, in relation to risks that cannot be eliminated;
- c) Provide information for safety (warnings/precautions/contra-indications) and, where appropriate, training to users.
For security, a similar approach can be taken:
a) Eliminate or reduce security risks as far as feasible through secure design and manufacture;
b) Where appropriate, take adequate protection measures, including security notifications if necessary, in relation to risks that cannot be eliminated;
c) Provide information for security (warnings/precautions/contra-indications) including information on measures that the user is required to take in the operating environment to reduce the likelihood of exploitation.
When determining security capabilities, the manufacturer should demonstrate for each security measure that not only the goals of safety and effectiveness are maintained with the implementation of a specific capability, but also performance requirements and the existing risk control measures remain effective as specified.
3.4. Security Risk Assessment
When choosing such security capabilities as protection measures, the manufacturer should consider the device's intended clinical use and intended operational environment when determining the appropriate balance of safety, effectiveness and security. Threat Modelling techniques are a systematic approach for analysing the security of an item in a structural way such that vulnerabilities can be identified, enumerated, and prioritised, all from a hypothetical attacker's point of view. Threat modelling can be applied to software, devices, systems, networks, distributed systems, business processes, etc. Threat modelling typically employs a systematic approach to identify attack vectors and assets most desired by an attacker. This leads to a decomposition of the item (software, device, system, etc.) to look at each possible attack vector and asset individually and determine to which kind of attacks they are vulnerable. From this, a list of vulnerabilities can be created and ordered in terms of risk, potential to affect safety and effectiveness, or any other criteria deemed appropriate.
Note: Many vulnerabilities exist, most of which are unknown (in the sense that nobody has ever identified a potential threat scenario for that vulnerability). An identified vulnerability is typically identified via a Common Vulnerabilities and Exposures (CVE) identifier. A scenario whereby an attacker exploits a known vulnerability may be considered as 'foreseeable' with respect to the product's risk management - notably if the intended operational environment or other mitigation controls do not prevent that type of attack.
Note: The likelihood of identified security scenarios can be supported through structured scoring systems, e.g. Common Vulnerability Scoring Systems - CVSS 13 , which may also take into account the attacker's gain in relation to the required effort.
3.5. Security Benefit Risk Analysis
Carrying out a Benefit Risk Analysis is an explicit requirement of the Medical Devices Regulations Annex I, sections 1, 2, 3e and 8. It shall be noted that the Benefit Risk Analysis is not executed for every individual security risk. Instead, an overall Benefit Risk Analysis is to be executed based on the intended use and possible safety and performance impact using the safety risk assessment, which includes the security-related hazard categories (as defined in chapter 3.2 Security Risk Management).
Risk acceptance criteria should be established by the manufacturer and documented to guide the appropriate measures for mitigating security risks. Those criteria relate to the intended purpose and operational environment.
3.6. Minimum IT Requirements
Annex I of the Medical Devices Regulations make explicit references to the environment hosting medical devices, highlighting the need for medical device manufacturers to set out the minimum relevant IT security requirements (17.4 MDR/16.4 IVDR) and communicate them effectively to the users (23.4ab MDR/20.4 ah IVDR):
[17.4 MDR/16.4 IVDR] Manufacturers shall set out minimum requirements concerning hardware, IT networks characteristics and IT security measures, including protection against unauthorised access, necessary to run the software as intended
As regards to the operating environment, this section should be interpreted as follows:
- It is the manufacturers' responsibility to determine the minimum requirements for the operating environment as regards IT network characteristics and IT security measures that could not be implemented through the product design.
- IT security measures may refer to any applicable technical and/or organisational measures for managing IT security risks related to the operating environment.
[23.4 (ab) MDR/20.4 (ah )IVDR] Information in the instructions for use
The instructions for use shall contain all of the following particulars: for devices that incorporate electronic programmable systems, including software, or software that are devices in themselves, minimum requirements concerning hardware, IT networks characteristics and IT security measures, including protection against unauthorised access, necessary to run the software as intended.
This section should be interpreted as follows:
- The manufacturer shall provide clear documentation of the device's instructions for use, including IT security features/configurations (if applicable), and clear instructions for the IT security controls related to the operating environment, including product specifications, compatibilities, recommended IT security measures, IT environment configuration (e.g. traffic control), etc.
Due to frequent changes in the threat landscape, it might be advisable to maintain security information in an electronic form that allows for dynamic updates as needed.
Basic principles
The operating environment for a medical device is defined as any IT/network asset interacting with the medical device that is not supplied by the medical device manufacturer.
Any minimum requirements concerning hardware, IT networks characteristics and IT security measures for the operating environment should be defined on the basis of the following principles:
Any proposed IT security requirement for the operating environment should be based on the risk assessment conducted for the medical device.
The medical device should be as autonomous as possible in terms of IT security and sole reliance on the existence of any IT security requirements on the operating environment should be kept to a minimum and reflect the manufacturer's assumptions on the baseline environment security for the secure operation of the medical device.
The manufacturer's assumptions regarding the IT security of the operating environment shall be clearly documented in the instructions for use and may refer to best practice security standards.
In accordance with the principle of layered security, IT security measures foreseen for the operating environment in general should not serve the purpose of compensating security controls for medical device vulnerabilities, unless there is sufficient justification. In cases where the medical device relies on the operating environment to provide important IT security controls, this should be stated in the accompanying technical documentation.
IT security requirements for the operating environment
The medical device manufacturer should determine the IT security requirements for the operating environment on the basis of the aforementioned principles. The relevant security requirements may include any combination of technical and organisational measures that affect the IT security of the operating environment of the medical device. The operating environment is defined as the sum of IT assets (software, hardware, network components) within which the medical device operates and with which the medical device interacts.
The security measures listed below should be viewed as a non-exhaustive and non-mandatory list of possible security controls for the operating environment. Moreover, they include IT security practices that are beneficial for the overall IT security posture of the operator's IT environment (good practices) but may not necessarily be considered mandatory as regards to the suitability of the operating environment.
General security requirements for operating environment
The following indicative list of IT security requirements is suggested for the operating environment of medical devices. The exact requirements should be defined by the medical device manufacturer on a per case basis, since not all security measures are systematically applicable in all contexts.
The operator must be in line with national and EU regulations (e.g. GDPR).
The operating environment must provide physical security for the medical device via security measures such as:
Regulated and authenticated physical access enforced via suitable technical measures (e.g. badges)
Physical security policy defining roles and access rights, including for physical access to the medical device
Use of segregated, secure areas with appropriate access controls
The operating environment must include appropriate security controls such as:
User access management (credentials for accessing software applications or devices, user access policy, etc.)
Antivirus / anti-malware software
Firewall
Application whitelisting / system hardening
Exclusive use of genuine software and ban of all illegitimate software and applications
Session management measures (e.g. session timeouts)
The operating environment must provide control and security of network traffic via appropriate measures, such as:
Network segmentation
Traffic filtering
Data encryption
Specifically for the workstations connected to the medical device, appropriate security measures may include:
Operating system hardening and application whitelisting
Memory protection measures to block arbitrary code execution
Compatibility of medical device management software with security solutions that counter malicious code
Use of strong passwords
Install only software programmes necessary for the intended use of the operating environment.
For cases when the operating environment is a complex system integrating multiple medical devices and other systems, appropriate measures to limit the propagation of an attack may include:
Partitioning mechanisms and network / traffic segmentation
Software integrity checks and device authentication mechanisms
To ensure that the security posture of the operating environment and of the device itself remain at a suitable level, appropriate provisions regarding patch management should be in place, such as:
The operating environment should support patching without compromising interoperability/compatibility
The operator should have appropriate patch management processes to ensure that security patches for medical devices are deployed in a timely manner
The operator should have appropriate patch management processes to ensure that the operating environment (e.g. operating systems, applications) is up-to-date in terms of security
Elements of the operating environment interacting with (e.g. other devices) or required for the operation of medical devices (e.g. OS) should ensure interoperability and shall not impair the specified performance of the medical device 14
3.7. Verification/Validation
MDR Annex I Section 17.2 and IVDR Annex I Section 16.2 require for devices that incorporate software or for software that are devices in themselves, that the software shall be developed and manufactured in accordance with the state of the art taking into account the principles of the development life cycle, risk management, including information security, verification and validation.
The primary means of security verification and validation is testing. Methods can include security feature testing, fuzz testing, vulnerability scanning and penetration testing 15 16 . Additional security testing can be done by using tools for secure code analysis and tools that scan for open source code and libraries used in the product, to identify components with known issues.
3.8. Lifecycle Aspects
Addressing cybersecurity risks at the design stage can help mitigate cybersecurity risks that could contribute to a breach in the confidentiality, a compromise in the integrity and availability of the medical device and its data, or intentional unauthorised access to the medical device and/or the network. Compromised CIA might impact medical purposes as specified in the medical device definition in MDR Article 2.
Other than for safety related hazards, whose number is quite stable over time, the security situation for software may change rapidly due to newly emerging security vulnerabilities, or due to new attack vectors.
This may lead to the situation that a medical device is considered secure with respect to known vulnerabilities at a specific point in time. However, ithout any security maintenance that device may become unsecure and possibly unsafe as a consequence due to newly emerging vulnerabilities or due to novel attack methods.
During the support lifetime of the device, the manufacturer should put in place a process to gather post-market information with respect to the security of the device (see also Chapter 6). This process should take into account:
- Security incidents directly related to medical device software
- Security Vulnerabilities that are related to the medical device hardware/software and the 3rd party hardware/software used with the medical device.
- Changes in the threat landscape, including interoperability aspects
The manufacturer should evaluate the information thus gathered, evaluate the associated security and safety risk and take appropriate measures that control the risk associated with such security incidents or vulnerabilities. Measures may include:
- Information to operators of medical devices on the identified risk and possible mitigations in the operating environment
- Quick fixes, e.g. network configuration changes
- Medical device software updates
- 3rd party software updates or patches.
The measures should be implemented at the operator site in a time appropriate to the security and safety risk determined by the manufacturer and operator.
4. Documentation and Instructions for use
4.1. Documentation
Medical Devices Regulations Annex I, section 3 explicitly requests from manufacturers to establish, implement, document and maintain a risk management system (see Chapter 3.2). The requirements for such documentation are described in Medical Devices Regulations Article 10 according to which manufacturers of devices other than custom-made devices shall draw up and keep up to date technical documentation for those devices. Overall, the technical documentation shall include the elements set out in Medical Devices Regulations Annexes II (technical documentation) and III (technical documentation on post-market surveillance) 17 .
According to Annex II of the Medical Devices Regulations, documentation shall contain information for the demonstration of conformity with the general safety and performance requirements set out in Medical Devices Regulations Annex I. These include security requirements to ensure safety and effectiveness of products against security risks and threats (see Chapter 2.3 of this guidance), and shall comprise a justification, validation and verification of the solutions adopted to meet those requirements (e.g. methods and results of security testing described in Chapter 3.7 of this guidance). In addition, the technical documentation needs to be updated with information raised through the manufacturers post market surveillance system related to handling and remediation of cybersecurity incidents and vulnerabilities (see Chapter 6 of the present guidance and Medical Devices Regulations Annex III).
4.2. Instructions for use
The requirements regarding the instructions for use are outlined in the following articles of Annex I:
[23.4 MDR/ 20 IVDR] Information in the instructions for use
'The instructions for use shall contain all of the following particulars:
'(...)
23.4 (g) MDR any residual risks, contra-indications and any undesirable side-effects, including information to be conveyed to the patient in this regard;'
20.1 (g) IVDR Residual risks which are required to be communicated to the user and/or other person shall be included as limitations, contra-indications, precautions or warnings in the information supplied by the manufacturer.
In the context of cybersecurity, this article sets out the need to provide information on the risk assessment for the device as regards to IT security risks. In this context, relevant information could include:
- High level summary of risk profile of the medical device and the corresponding IT security objectives (e.g. processing/protection of sensitive information, requirement for uninterrupted operation etc.)
'(h) specifications the user requires to use the device appropriately, e.g. if the device has a measuring function, the degree of accuracy claimed for it;'
Information that could be provided in this context includes aspects such as medical device IT characteristics, such as:
Specifications of the operating system
Provisions to ensure integrity/validation of software updates and security patches
1 rev 8 available at https://ec.europa.eu/docsroom/documents/32305
23.4 (i) MDR') details of any preparatory treatment or handling of the device before it is ready for use or during its use, such as sterilisation, final assembly, calibration, etc., including the levels of disinfection required to ensure patient safety and all available methods for achieving those levels of disinfection; '
20.4.1 (r) IVDR 'details of any preparatory treatment or handling of the device before it is ready for use, such as sterilisation, final assembly, calibration, etc., for the device to be used as intended by the manufacturer;'
In the context of cybersecurity, this requirement should be interpreted as referring to installation, configuration and operation of the medical device. Information that could be provided in this context includes:
- Security configuration options; in accordance with the security-by-default principle, the medical device should have the highest possible security settings selected by default.
- Product installation
- Initial configuration guidelines, e.g. change of default passwords during first login.
- Step-by-step instructions for deploying security updates
- Procedures for using the medical device in failsafe mode (e.g. enter/exit failsafe mode, performance restrictions in failsafe mode, data recovery function when resuming normal operation etc.)
- Documented action plan for the user to follow in case of an alert message
23.4 (j) MDR 'any requirements for special facilities, or special training, or particular qualifications of the device user and/or other persons;'
20.4.1(p) IVDR 'where relevant, requirements for special facilities, such as a clean room environment, or special training, such as on radiation safety, or particular qualifications of the intended user;'
In the context of cybersecurity requirements information that could be provided includes:
- User requirements in terms of training / required skills, including IT skills required for the installation, configuration and operation of the medical device
23.4 (k) MDR and 2.4.1 (s) IVDR 'the information needed to verify whether the device is properly installed and is ready to perform safely and as intended by the manufacturer, together with, where relevant:
- details of the nature, and frequency, of preventive and regular maintenance, and of any preparatory cleaning or disinfection,
- methods for eliminating the risks encountered by persons involved in installing, calibrating or servicing devices;'
In the context of cybersecurity, information relevant to this article involves the secure configuration and application of security updates for the medical device, e.g.:
- Provisions to ensure integrity/validation of software updates and security patches
- Step-by-step instructions for deploying security updates
23.4 (q) MDR 'for devices intended for use together with other devices and/or general purpose equipment:
- information to identify such devices or equipment, in order to obtain a safe combination, and/or
- information on any known restrictions to combinations of devices and equipment;'
20.4.1 (j) IVDR 'for devices intended for use in combination with or installed with or connected to other devices and/or general purpose equipment:
- information to identify such devices or equipment, in order to obtain a validated and safe combination, including key performance characteristics, and/or
- information on any known restrictions to combinations of devices and equipment.'
In the context of cybersecurity, this article sets out the requirement to outline compatibility issues as regards to the operating environment (software, hardware etc.) and any compatibility restrictions. Information that could be provided in this context includes:
- Minimum requirements for the workstations intended for user operations: hardware features, operating system versions, peripheral devices, etc.
23.4 (ab) MDR and 20.4.1 (ah) IVDR 'for devices that incorporate electronic programmable systems, including software, or software that are devices in themselves, minimum requirements concerning hardware, IT networks characteristics and IT security measures, including protection against unauthorised access, necessary to run the software as intended.'
This article sets out the need for the instructions for use to adequately describe the requirements regarding the operating environment (hardware, network characteristics, security controls etc.). Information that could be provided in this context includes:
- Assumptions on the environment of use (e.g. home environment, healthcare facility etc.)
- Risks for device operation outside the intended operating environment
- Minimum platform requirements for the connected medical device: hardware properties, operating system versions, middleware and drivers, peripheral devices, etc.
- Recommended IT security controls for operating environment (e.g. anti-virus, firewall)
- A description of backup and restore features for both data and configuration settings
Often, specific security information is shared through documentation other than the instructions for use, such as instructions for administrators or security operation manuals. Such information may include the following:
List of IT security controls included in the medical device
Depending on the type of product, provisions to ensure integrity/validation of software updates and security patches
Technical properties of hardware components
Software Bill of Materials
User roles and respective access privileges/permissions on the device
Implementation of the logging function, particularly the medical device's log storage capacity and the recommendations for backing up and using the logs
Launch of production system including guidelines on security recommendations and requirements relating to integration of the medical device within a health information system
System operation, administration, monitoring and operation support
Minimum requirements for the administration workstation for the connected medical device: hardware properties, operating system versions, middleware and drivers, peripheral devices, etc.
In case of network-connected medical devices, the documentation should contain an exhaustive matrix of the network data streams (protocol types, origin/destination of data streams, addressing scheme, etc.)
If the operating environment is not exclusively local but involves external hosting providers, the documentation must clearly state what, where and how data is stored, as well as any security controls to safeguard the data in the cloud environment (e.g. encryption)
Specific configuration requirements for the operating environment, such as firewall rules (ports, interfaces, protocols, addressing schemes etc.)
4.3. Information to be provided to healthcare providers
Among the information to be provided to healthcare providers regarding the intended use environment, the following can be included (non-exhaustive list):
- Device instructions for use and product specifications related to recommended cybersecurity controls appropriate for the intended use environment (e.g., anti-virus software, use of a firewall, etc.).
- Description of device features that protect critical functionality, even when the device's cybersecurity has been compromised (e.g. Operating System hardening).
- Description of backup and restore features and procedures to regain configurations.
- Specific guidance to users regarding supporting infrastructure requirements so that the device can operate as intended.
- Description of how the device is or can be hardened using secure configuration. Secure configurations may include end point protections such as anti-malware, firewall/firewall rules, whitelisting, security event parameters, logging parameters, physical security detection, etc.
- List of network ports and other interfaces that are expected to receive/send data, and a description of port functionality and whether the ports are incoming or outgoing (Unused ports should be disabled).
- Sufficiently detailed network diagrams for end-users.
- Where appropriate, technical instructions to permit secure network (connected) deployment and servicing, and instructions for users on how to respond upon detection of a cybersecurity vulnerability or incident.
- A description of how the design enables the device to announce when anomalous conditions are detected (i.e., security events). Security event types could be configuration changes, network anomalies, login attempts, anomalous traffic (e.g., send requests to unknown entities).
- A description of how forensic evidence is captured, including but not limited to any log files kept for a security event. Log files descriptions should include how and where the log file is located, stored, recycled, archived, and how it could be consumed by automated analysis software (e.g., Intrusion Detection System, IDS).
- A description of the methods for retention and recovery of device configuration by an authenticated privileged user.
- Where appropriate, risks of using the medical device outside of the intended use environment.
The MDS2 is an industry-wide and globally accepted form, which can be used to provide the abovementioned security information.
Clinicians/physicians should be provided with the information they need to have meaningful discussion with their patients about the risks and benefits of the device they use, including cybersecurity risks.
Provision of information for Medical Device Software (MDSW) users should be tailored to where the device is used (e.g. home environment with limited cyber protection, public environment).
5. Post-Market Surveillance and Vigilance
The post-market phase of medical devices life cycle is a crucial aspect that manufacturers shall implement as cybersecurity vulnerabilities change and evolve over time and controls implemented during pre-market activities may be inadequate to maintain an acceptable benefit-risk level.
An effective and successful post-market cybersecurity surveillance program should include the following aspects:
- operation of the device in the intended environment
- sharing and dissemination of cybersecurity information and knowledge of cybersecurity vulnerabilities and threats across multiple sectors
- vulnerability remediation
- incident 18 response
5.1. Post-market surveillance system
The manufacturer is required to put in place a post market surveillance (PMS) system and actively keep this PMS up to date in accordance with MDR Art. 83 or IVDR Art. 78. Cybersecurity considerations for medical devices should be part of this PMS system. A PMS system includes actively and regularly collecting user experience from devices on the market (including third party software and hardware components), to review these, and to timely implement necessary corrective action, taking into account the nature and risks in relation to the device. The manufacturer will involve the distributors of the device and, where applicable, the authorised representative and importers of the device in his system, in order to obtain the relevant information from the market.
This system will be part of QMS, and be supported by the manufacturer's PMS plan 19 , which must address a range of information (Medical Devices Regulations Annex III).
According to the Medical Devices Regulations, an incident 20 is any malfunction or deterioration in the characteristics or performance of a device made available on the market, including use-error due to ergonomic features, as well as any inadequacy in the information supplied by the manufacturer and any undesirable side-effect. While a serious incident 21 is defined as any incident that directly or indirectly led, might have led or might lead to any of the following:
- the death of a patient, user or other person,
- the temporary or permanent serious deterioration of a patient's, user's or other person's state of health,
- a serious public health threat
A few examples are provided in Annex II of this guidance on the distinction between incidents and serious incidents from the point of view of cybersecurity. In this table, in respect to serious incidents, all the foreseen control measures listed, security control and safety control that are needed to be implemented in order to eliminate or reduce the risk of patient harm (safety harm) are considered examples of FSCA.
Depending on the class of the device, a PMS report 22 or a PSUR report 23 will be prepared, that summarises the results and conclusions of the analysis of all the data from the market.
Data gathered from PMS system must be used to actively update:
- the clinical evaluation;
- the benefit-risk determination and to improve the risk management;
- the design and manufacturing information, the instructions for use and the labelling;
Handling and remediation of cybersecurity incidents and vulnerabilities reported through the postmarket surveillance and vigilance systems shall be carried out conforming to the methodologies described in Chapter 3.2 of this guidance, with regards to:
- Assess the need for reporting serious and non-serious incidents and of carrying-out field safety corrective actions;
- Enhancing security capabilities;
- Update the original Security Risk Assessment;
- Update the Verification and Validation;
- Update the original Security Benefit Risk Analysis
- Update the Technical Documentation.
5.2. Vigilance 24
The manufacturer is responsible for reporting all serious incidents and field safety corrective actions (FSCA) to the CA in accordance with MDR Article 87 or IVDR art 82. According to the Medical Devices Regulations, field safety corrective action' means corrective action taken by a manufacturer for technical or medical reasons to prevent or reduce the risk of a serious incident in relation to a
Medical Device Coordination Group Document device made available on the market. Manufacturers are obliged to conduct investigations as soon as they are informed that a serious incident has taken place. As such, a risk assessment of the incident is conducted and if needed, a FSCA will be implemented in order to minimize the risk of the device.
The manufacturer will involve the distributors of the device and, where applicable, the authorised representative and importers in the system, in order to obtain the information needed from the market, especially for FSCA or issued field safety notices (FSN) so that to ensure required actions are followed and completed in a timely manner.
The manufacturers shall carry out investigations of serious incidents related to a cybersecurity incident in order to provide a comprehensive description of the serious incident, including:
- a) a description of the serious incident including any relevant information that might impact the understanding or evaluation of the serious incident, i.e. information is compromised or information is threatened;
- b) a description of the health effects (if applicable), i.e. clinical signs, symptoms, conditions as well as the overall health impact.
The reporting tools that are made available to manufacturer enable the use of IMDRF codes to index:
- the device problem of the incident;
- the related health impact;
- cybersecurity related incident root causes;
Regarding medical device problems due to cybersecurity related incident root causes, the IMDRF codes available to date include: code A110502 (IMDRF Annex A), which is representative for a Computer System Security Problem. This code is further sub-divided into 2 more specific codes on "Application Security Problem" and "Unauthorized Access to Computer System" (see Table 4). Regarding the cybersecurity related incident root causes of medical device problems, the IMDRF codes available to date include code C1007 (IMDRF Annex C) which refers to "Software Security Vulnerability" (see Table 5).
Table 4 : IMDRF Annex A codes on Cybersecurity related device problems
| Level 2 | Level 2 | Level 2 | Level3 | Level3 | Level3 |
|---|---|---|---|---|---|
| Term | Definition | Code | Term | Definition | Code |
| Computer System Security Problem | Problem associated with unauthorized access to or modification of a software system resulting in a loss of confidentiality, integrity, or availability of written program code, application software, or data or entire device. | A1105 | Application Security Problem | Problem associated with the acquisition of computer programming codes that can replicate and spread from one computer system to another thereby leading to damaged software, hardware and data. | A110501 |
| Computer System Security Problem | Problem associated with unauthorized access to or modification of a software system resulting in a loss of confidentiality, integrity, or availability of written program code, application software, or data or entire device. | A1105 | Unauthorized Access to Computer System | Problem associated with an access that was not permitted to the computer system that may lead to modification of program, corruption of data, or and break in network security. This concept is closely associated with computer integrity which is the degree to which a system or component prevents unauthorized access to, or modification of, computer programs or data. | A110502 |
Table 5: IMDRF Annex C codes on Cybersecurity related incident root causes
| Level3 | ||
|---|---|---|
| Term | Definition | Code |
| Software Security Vulnerability | The device software failed to provide adequate authorization, access control, protection and accountability features. | C1007 |
In cases where the currently available IMDRF coding system does not provide sufficient level of detail to indicate the cybersecurity causal root of an incident, the manufacturer is requested to propose a new code to be adopted by the IMDRF coding system, in accordance with the relevant procedure for new term request 25 .
For similar serious incidents that occur with the same device or device type and for which the root cause has been identified or a field safety corrective action implemented or where the incidents are common and well documented, the manufacturer may provide periodic summary reports (PSR) instead of individual serious incident reports. This, however, is permissible only when the coordinating competent authority, has agreed with the manufacturer on the format, content and frequency of the periodic summary reporting 26 .
Incidents that have cybersecurity related incident root causes are subject to Trend Reporting under the Medical Devices Regulations 27 . According to trend reporting provisions, manufacturers shall report, by means of the electronic system referred to in Article 92 28 , any statistically significant increase in the frequency or severity of incidents that are not serious incidents or that are expected undesirable side-effects that could have a significant impact on the benefit-risk analysis referred to in Sections 1 and 8 of Annex I and which have led or may lead to risks to the health or safety of patients, users or other persons that are unacceptable when weighed against the intended benefits.
Within the Trend Report, key obligations of the manufacturers are to specify:
- the methodology used for determining any statistically significant increase in the frequency or severity;
- how to manage the incidents;
- the observation period.
Using IMDRF codes to index the cybersecurity medical root causes related to non-serious incidents is desirable and may be implemented into the Trend Report 29,30 .
6. Other Legislation and guidance: EU and International
6.1. EU Legislation in the sector
At EU level, the following legislative acts are relevant to the cybersecurity of medical devices or to operators dealing with protecting or processing of personal data stored in medical devices and might apply in parallel to the Medical Devices Regulations:
- NIS Directive 31
- GDPR (General Data Protection Regulation) 32
The NIS Directive provides legal measures to boost the overall level of cybersecurity in the EU by ensuring:
- Member States preparedness by requiring them to be appropriately equipped, e.g. via a Computer Security Incident Response Team (CSIRT) and a competent national NIS authority,
- Cooperation among all the Member States, by setting up a cooperation group, in order to support and facilitate strategic cooperation and the exchange of information among Member States. They will also need to set a CSIRT Network, in order to promote swift and effective operational cooperation on specific cybersecurity incidents and sharing information about risks,
- A culture of security across sectors which are vital for our economy and society and moreover rely heavily on ICT, such as energy, transport, water, banking, financial market infrastructures, healthcare and digital infrastructure. Businesses in these sectors that are identified by the Member States as operators of essential services will have to take appropriate security measures and to notify incidents of significant impact to the relevant national authority. Also, key digital service providers (search engines, cloud computing services and online marketplaces) will have to comply with the security and notification requirements under the new Directive.
The General Data Protection Regulation ('GDPR') regulates the processing by an individual, a company or an organisation of personal data relating to individuals in the EU. Personal data is any information that relates to an identified or identifiable living individual . Different pieces of information, which collected together can lead to the identification of a particular person, also constitute personal data. Personal data that has been de-identified, encrypted or pseudonymised but can be used to re-identify a person remains personal data and falls within the scope of the GDPR. Personal data that has been rendered anonymous in such a way that the individual is not or no longer identifiable are no longer considered personal data. For data to be truly anonymised, the anonymization must be irreversible.
The GDPR protects personal data regardless of the technology used for processing that data - it's technology neutral and applies to both automated and manual processing, provided the data is organised in accordance with pre-defined criteria (for example alphabetical order). It also doesn't matter how the data is stored - in an IT system, through video surveillance, or on paper; in all cases, personal data is subject to the protection requirements set out in the GDPR.
In Europe, guidance documents were developed by France 33 , Germany 34 , Switzerland 35 and the UK, moreover other national legislation could apply (e.g. ASIP certification in France, NEN 7510 in the Netherlands). The principles therein contained are incorporated into this guidance document.
Finally at EU level, must be mentioned the EU Cybersecurity Act 36 that introduces for the first time an EU-wide cybersecurity certification framework for ICT products, services and processes.
6.2. IMDRF Guide on Cybersecurity of Medical Devices
At worldwide level, it is important to refer the Medical Device Cybersecurity Guide 37 under development by a Working Group of the International Medical Device Regulators Forum (IMDRF). The purpose of this work item is to promote a globally harmonized approach to medical device cybersecurity and to provide medical device cybersecurity guidance for stakeholders across the device lifecycle.
cybersicherheit.de/ACS/DE/_/downloads/BSI-CS_132E.pdf?__blob=publicationFile&v=5
7. Annex I - Mapping of IT security requirements to NIS Directive Cooperation Group measures
IT security requirements for the operating environment
In addition to the requirements described in Chapter 3.6 of this guidance, operators should also adopt good practices as regards to cybersecurity to achieve a good cybersecurity standing with a direct impact on the security of the medical device. Indicative examples of such practices include:
- Conduct a risk assessment and impact assessment. Introduction of medical devices in the environment should be subject to such a risk assessment.
- A set of baseline IT security policies should be defined, approved by management and communicated to employees and relevant external parties. Examples of such policies include:
- Define clear roles for users of critical systems and medical devices, segregate duties
- Define information classification levels and label devices handling such information accordingly
- Acceptable use policy
- Password policy
- Change Management policy
- Establish a backup policy on selected critical systems.
- Establish a vendor/procurement policy for the provision of medical equipment.
- Include proper SLAs and NDAs to vendors and external associates
- Provide security awareness training for employees that operate critical devices and systems and make background checks prior to authorizing access to key personnel.
- Catalogue assets in an inventory of all medical devices , servers and workstations.
- Assign unique ID's to healthcare information systems, medical devices.
- Monitor and keep track of changes in ecosystem parties, so that business processes are not interrupted or hide risks.
- Apply the principle of least privilege to user workstations and connected devices.
- Least privileges must also take into account data minimisation per role.
- Data integrity should be ensured e.g. through hashing, integrity checks.
- Establish appropriate security measures for the use of mobile devices and teleworking .
- The Health Information System (HIS) must be able to monitor the correct operation of the equipment .
- Monitor device behaviour in the context of medical workflows.
- Implement data recovery mechanisms to restore data from critical systems
- Investigate major incidents and review actions taken to mitigate and reduce time to react to future occurrences.
- Develop a disaster recovery plan , taking into account the minimum recovery requirements.
- Avoid the use of End of life third-party components and devices on the operating environment, where possible take additional measures such as network isolation.
The aforementioned general security requirements for the operating environment along with the minimum IT requirements listed in chapter 3.6 are tabulated below, mapped to the security measures for OES published by the NIS Directive Cooperation Group. The minimum IT security requirements for the operating environment are listed in bold , whereas the general good practices in regular font.
Particular emphasis should be placed on the fact that, upon implementation of the Directive on security of network and information systems (NIS Directive) 38 , many operators 39 will be obligated to introduce IT security measures in their respective IT environments. The following table provides a mapping of security measures to the measures defined in the 'Reference document on security measures for Operators of Essential Services' published by the NIS Directive Cooperation Group 40 . Still, the following should be noted:
- Not all operators will be identified as Operators of Essential Services (OES) and, as such, be subject to the provisions of the NIS Directive as regards security measures.
- Member States will define their own security measures for OES in the healthcare sector. The measures proposed by the Cooperation Group are non-binding and serve as guidelines for Member States.
| D/N | DOMAIN NAME | SECURITY CATEGORY | MEASURES |
|---|---|---|---|
| Part 1 - Governance and Ecosystem | |||
| 1.1 | Information System Security Governance &Risk Management | Information system security risk analysis | Conduct a risk assessment and impact assessment based on industry standards (like DICOM or ISO 80001 for medical devices or ISO 27799). Medical device introduction should be subject to such a risk assessment |
| 1.1 | Information System Security Governance &Risk Management | Information system security policy | Adequate patch management processes o for medical devices o for the operating environment o Deployed in a timely manner Only use genuine software and ban all illegitimate software Define a set of baseline IT security policies o Management should approve the policies o Define clear roles for users of critical systems and medical devices, segregate duties o Define information classification levels and label devices handling such information accordingly o Acceptable use policy o Change Management policy o Establish a backup policy on selected critical systems. o Establish a vendor/procurement policy for the provision of medical equipment. o Include proper SLAs and NDAs to vendors and external associates Policies communicated to employees and external parties |
| 1.1 | Information System Security Governance &Risk Management | Information system security accreditation | |
| 1.1 | Information System Security Governance &Risk Management | Information system |
| D/N | DOMAIN NAME | SECURITY CATEGORY | MEASURES |
|---|---|---|---|
| security indicators | |||
| Information system security audit | |||
| Human resource security | Security awareness training for employees that operate critical devices and systems Background checks prior to authorizing access to key personnel | ||
| Asset Management | Catalogue medical devices and relevant assets in inventory. o Assign unique ID's to healthcare information systems, medical devices | ||
| 1.2 | Ecosystem Management | Ecosystem mapping | |
| 1.2 | Ecosystem Management | Ecosystem relations | |
| Part 2 - Protection | |||
| 2.1 | IT Security Architecture | Systems configuration | Operating environment must not hinder the application of security measures on the medical device or force the device to operate in lower security settings. Session management measures (e.g. session timeouts). Operating system hardening and application whitelisting Antivirus / anti-malware software Use of strong passwords Appropriate security measures for mobile devices and teleworking |
| 2.1 | IT Security Architecture | System segregation | Firewall Network segmentation Partitioning mechanisms and traffic segmentation |
| 2.1 | IT Security Architecture | Traffic filtering | Use of traffic filtering software and hardware |
| 2.1 | IT Security Architecture | Cryptography | Encryption when storing sensitive personal data Encryption of data in transit |
| 2.2 | IT Security Administration | Administration accounts | |
| 2.2 | IT Security Administration | Administration information systems | Memory protection measures to block arbitrary code execution Compatibility of medical device management software with security solutions that counter malicious code. Install only software programmes necessary for intended uses . |
| 2.3 | Identity and access management | Authentication and identification | User access management (credentials for accessing software applications or devices, user access policy etc. ) |
| 2.3 | Identity and access management | Access rights | Apply principle of least privilege to user workstations and connected devices. Least privileges must take into account data minimisation per role |
| 2.4 | IT security maintenance | IT security maintenance | Provisions regarding patch management Support patching without compromising interoperability/compatibility |
| D/N | DOMAIN NAME | SECURITY CATEGORY | MEASURES |
|---|---|---|---|
| procedure | Do not use End of life third-party components and devices on the operating environment | ||
| Remote access | |||
| 2.5 | Physical and environmental security | Physical and environmental security | Regulated and authenticated physical access via suitable technical measures (e.g. badges) Define roles and access rights, including those for physical access to medical devices Use of segregated, secure areas with appropriate access controls |
| Part 3 - Defence | |||
| 3.1 | Detection | Detection | Software integrity checks and device authentication mechanisms Data integrity ensured e.g. through hashing, integrity checks. |
| 3.1 | Detection | Logging | Monitor and keep track of changes in ecosystem parties, so that business processes are not interrupted or hide risks |
| 3.1 | Detection | Logs correlation and analysis | The Health Information System (HIS) must be able to monitor the correct operation of the equipment. o Monitor device behaviour in the context of medical workflows |
| 3.2 | Computer security incident management | Information system security incident response | |
| 3.2 | Computer security incident management | Incident report | |
| 3.2 | Computer security incident management | Communication with competent authorities | |
| Part 4 - Resilience | |||
| 4.1 | Continuity of Operations | Business continuity management | Investigate major incidents and review actions taken to mitigate and reduce time to react to future occurrences |
| 4.1 | Continuity of Operations | Disaster recovery management | Develop a disaster recovery plan, taking into account the minimum recovery requirements Implement data recovery mechanisms to critical systems |
| 4.2 | Crisis Management | Crisis management organization | |
| 4.2 | Crisis Management | Crisis management process |
8. Annex II - Examples of cybersecurity incidents/serious incidents
The examples included in this annex provide a representation of the relationship between cybersecurity risk management and patient safety management.
Disclaimer: the table below is intended for illustrative purpose only. In particular some of the harms and controls listed should not be interpreted as being applicable only for the device types provided.
| Serious incident (Yes/No) | Security Harm | Safety Harm | ||
|---|---|---|---|---|
| Serious incident (Yes/No) | Security Control | Safety Control | ||
| Yes | Security risk with a safety impact. | External Programmer for an implantable Deep Brain Stimulator | Custom malware is installed on the External Programmer / Modification of External Programmer function, including stimulation parameters. | Increased, decreased, and/or an intermittent stimulation not intended in the current programming parameters; or, inability to change programs or control the amplitude using the patient |
| Yes | Security risk with a safety impact. | External Programmer for an implantable Deep Brain Stimulator | Establish message authentication between Programmer and IPG and Programmer prevents installation of third-party applications and limits access to the programmer device OS. | Not Applicable. |
| Yes | Security risk with a safety impact | External Programmer for an implantable Pacemaker | External Programmer is used by an unauthorized user to adjust therapy settings without the patient's knowledge. | Increased, decreased, and/or an intermittent stimulation not intended in the current programming parameters. |
| Yes | Security risk with a safety impact | External Programmer for an implantable Pacemaker | Implement User Authentication on External Programmer. | Inductive Programming Wand is required to start |
| communication session with the IPG (requires close patient proximity) | ||||
| Yes | Security risk with a safety impact | Implantable Sensor used to monitor Pulmonary Artery pressures in Heart Failure Patients | An attacker modifies or creates patient data in transit to or from the external electronics unit, causing misdiagnosis that affects patient care. | Physician fails to treat based on incorrect low PA pressure readings leading to worsening of patient's heart failure condition. |
| Yes | Security risk with a safety impact | Implantable Sensor used to monitor Pulmonary Artery pressures in Heart Failure Patients | Connection protocol from electronics unit to clinician website uses SSL/TLS encryption. | Not Applicable. |
| Yes | Security risk control with a safety impact. | Pacemaker | An unauthorized person is able to fatigue the device by overwhelming the device of requests. | A premature battery depletion may occur. |
| Yes | Security risk control with a safety impact. | Pacemaker | Avoid possibility to overwhelming the device. | Not Applicable. |
| Yes | Security risk control with a safety impact. | A smart infusion pump with its remote control. | Patient may reconfigure the device. | The smart infusion pump infuses more or less insulin than what was prescribed by an authorized user. |
| Yes | Security risk control with a safety impact. | A smart infusion pump with its remote control. | User type and access right should well be defined. | Not Applicable. |
| Yes | Security Risk within indirect safety impact (device availability) | Any Medical Device with Windows | Network-spread malware (worm) encrypts the content of the system hard drive. | No direct safety harm. (Indirect: MDnot available). |
| Yes | Security Risk within indirect safety impact (device availability) | Any Medical Device with Windows | Disconnect devices from network. | use of alternative devices. |
| Yes | Security risk with a safety impact. | Anaesthesia device | An unauthorized user with physical access to the | The anaesthesia device supplies a wrong anesthetic |
| device guesses the weak password for the service account and manipulates the configuration settings. | concentration. | |||
| Access control without password complexity enforcement. | Not Applicable. | |||
| No | Security risk only. | Warming therapy device for premature babies | An unauthorized user with physical access to the device guesses the weak password for the service account and exports therapy and patient data via the USB interface. | None. |
| No | Security risk only. | Warming therapy device for premature babies | Access control without password complexity enforcement. | Not Applicable. |
| Yes | Security risk with a safety impact. | Warming therapy device for premature babies | An attacker floods the network interface with tons of malformed service requests which causes the system to crash. | The therapy functionality of the device is not available. |
| Yes | Security risk with a safety impact. | Warming therapy device for premature babies | Not Applicable. | Not Applicable. |
| No | Security risk only. | Monitoring system | An attacker eavesdrop the network communication between a local patient monitor and the central monitoring station. Therefore the attacker gains possession of sensitive health information of the patient. | None. |
| No | Security risk only. | Monitoring system | Not Applicable. | Not Applicable. |
| Yes | Security risk with a safety impact. | Ventilator | An attacker with physical access installs malware on the device via the USB interface. | The respiration functionality of the device does not work as intended. |
| Yes | Security risk with a safety impact. | Ventilator | Not Applicable. | Not Applicable. |
| Yes | Security risk with a safety impact. | Monitoring system | An attacker with physical access to | Emergency measures are |
| the network manipulates a ventilator`s alarm messages sent to the central monitoring system. | not carried out in time. | |||
| Not Applicable. | ||||
| Yes | Security risk control with a safety impact. | PACS | An unauthorized user gains access to the local network and manipulates the network traffic between a device and the PACS Software. | There is the danger of manipulation of medical image data and thus the danger of false diagnoses. |
| Yes | Security risk control with a safety impact. | PACS | Networks Access Security. | User checks display data directly on device |
| Yes | Health damage caused by unavailability | PACS | An unauthorized user deploys malware (ransomware, scareware). | Health damage caused by unavailability. |
| Yes | Health damage caused by unavailability | PACS | Security Awareness Training, Firewall, Antivirus Solution, secure infrastructure, Backups. | User checks display data directly on device. |
| No | Security risk only. | PACS | Employee stealing data with mobile USB storage on a client pc. | None. |
| No | Security risk only. | PACS | Implement a User and Usergroup Permission Environment. | Not Applicable. |
| No | Delayed Treatment since system is not operational | MR | Network based attempted infection leading to system Blue Screen. | Since device is not available, Delayed treatment, meaning patient needs being scanned in another location or by other device. |
| No | No Impact, annoyance of the customer | MR | Network based infection, leading to contaminated system. System performs its functions, but slows down (at same time notifies | Slower performance in MRis a nuisance since all real time computing is not done on a machine which can be infected. |
| the operator). | ||||
| No | Security risk only | A Mobile X-ray C-arm with piggybacked network router | Unauthorized user changing router configuration via vulnerable management interface resulting in no network connectivity. | None. No impact on medical device and its essential performance. |
| No | Security risk only | A Mobile X-ray C-arm with piggybacked network router | Not Applicable. | Not Applicable. |
| Yes | Safety risk control with a security impact. | An x-ray machine. | Device vulnerable (e.g. RDP vulnerability) and network level communication attacked and disclosed due to delayed release and deployment of patches due to extensive V&V. | None. |
| Yes | Safety risk control with a security impact. | An x-ray machine. | Not applicable. | Full V&V of medical device and all controls before release. |
| Yes | Security risk with a safety impact. | An x-ray machine | System attacked and compromised through vulnerable legacy third party/network interfaces integrating with the medical device. | Delayed diagnosis and treatment due to unavailability. |
| Yes | Security risk with a safety impact. | An x-ray machine | Not applicable. | Not applicable. |
| Yes | Security risk control with a safety and security impact | An x-ray machine | DICOM objects infected with executable malware imported and exported spreading across PACS and medical device network. | Delayed diagnosis and treatment due to unavailability of compromised networked systems. |
| Yes | Security risk control with a safety and security impact | An x-ray machine | Hardening/Whitelisti ng blocking execution of DICOM objects. | Not applicable. |
| Yes | Security risk and control with a safety impact. | An interventional x- ray machine | Network attacked and compromised by worm or ransomware leading to shutdown of all systems not | Delayed diagnosis and treatment due to unavailability. |
| isolated and confirmed to be malware free. | ||||
| Hardened, locked down system blocking unvalidated malware scanning. | Not applicable. |
9. Annex III - Standards
Disclaimer: this section is intended for information purposes only and does not provide any guidance that could be used for the purposes of meeting requirements under Regulation (EU) 2017/745 or Regulation (EU) 2017/746. For example, standards referenced below cannot provide a presumption of conformity, unless they are harmonised under the aforementioned regulations and published in the Official Journal.
- EN ISO 14971 Risk Management (Product)
- EN 62304 Software Lifecycle
- EN ISO 31000 Risk Management (Organisation) or particular standards under ISO 31xxx.
- EN ISO/IEC 27000 Information technology - Security techniques - Information security management systems (ISMS) - Overview and vocabulary
- EN ISO/IEC 27001 Information Technology - Security techniques - Information Security management Systems - Requirements.
- EN ISO/IEC 60601-1-x
- IEC 82304-1 Health Software Part 1: General requirements for Product Safety
- ISO/IEC 80001-1 Application of Risk Management for IT networks Incorporating Medical Devices
- ISO/IEC 80001-5-1 Application of Risk Management for IT networks incorporating medical device - Safety, effectiveness and security in the implementation and use of connected medical devices or connected health software - Part 5-1: Activities in the product life-cycle.
- IEC/TR 80001-2-2 Application of Risk Management for IT networks Incorporating Medical Devices Part 2-2: Guidance for the Disclosure and Communication of Medical Device Security Needs, Risks and Controls
- IEC/TR 80001-2-8 Application of risk management for IT-networks incorporating medical devices - Part 2-8: Application guidance - Guidance on standards for establishing the security capabilities identified in IEC TR 80001-2-2
- ISO/IEC 80001-xx including IEC/TR 80001-2-1, IEC/TR 80001-2-3, IEC/TR 80001-2-4, IEC/TR 80001-2-5, ISO/TR 80001-2-6, ISO/TR 80001-2-7 or other
- EN ISO 62366 / ISO 60601-4 Usability Engineering
- IEC 62443-4-2 Security for industrial automation and control systems. Part 4-2: Technical security requirements for IACS components.
- IEC 62443-4-1 Security for industrial automation and control systems. Part 4-1: Secure product development lifecycle requirements.
- IEC/TR 60601-4-5 Medical Electrical Equipment - Part 4-5. Safety related technical security specifications for medical devices.
Cybersecurity Risk
Analysis
ISO 14971 Risk Management
Process
Risk Analysis
10. Annex IV - Cybersecurity risk management process and safety risk management relationship
The following figure illustrates the relationship between processes for cybersecurity risk management and safety risk management.
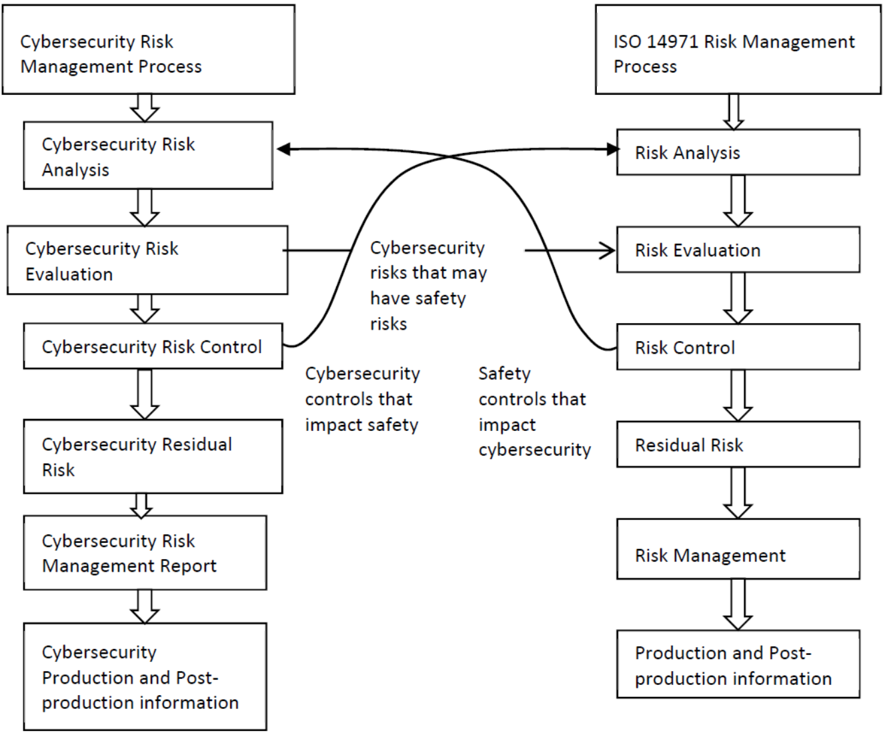
Footnotes
- Medical Device Directive (93/42/EEC), Directive on active implantable medical devices (90/385/EEC) and Directive on in vitro diagnostic medical devices (98/79/EC)
- Article 2 (23) of Regulation (EU) 2017/745 - MDR and Article 2(16) of Regulation (EU) 2017/746
- There is a need to distinguish between generic 'IT-networks' and 'medical IT-networks'
- Definition of Cybersecurity - Gaps and overlaps in standardisation (December 2015):
- idem
- idem
- MDR, Annex I Chapter 1, 3. (b)
- MDR Art. 7, Annex I, Chapter 1, Section 4
- For more information please visit:
- However, the EU believes that 'joint responsibility' is a more suitable term than 'shared responsibility' (the latter currently in use in the draft IDMRF document) to describe the separate expectations vis-a-vis the different stakeholders in the system (without prejudice to manufacturers' legal obligations and responsibilities laid down in the Regulation). The EU will recommend that the IMDRF adopt this term in the final version of their document, which is planned for 2020.
- Section 3 of MDR Annex I: establish and operate a risk management syste m across the entire lifecycle of the medical device as a continuous iterative process, requiring regular systematic updating.
- Annex I section 4 of the Medical Devices Regulations.
- Article 14.1, 14.2 and 14.5 of Annex I of the MDR addresses interactions between the medical device, the operating environment and other products as regards interoperability and performance
- According to ENISA, penetration testing is the assessment of the security of a system against different types of attacks performed by an authorised security expert. The tester attempts to identify and exploit the system's vulnerabilities.
- For further information on verification and validation, reference could be made to the guidance on clinical/performance evaluation of medical device software:
- See also Guidance document - Market surveillance - Guidelines on a Medical Devices Vigilance System - MEDDEV 2.12
- Whenever the word "incident" is used on its own in this guidance, it shall be understood as any incident that does meet the definition of "serious incident" as defined in the MD Regulations
- MDR Art. 84, IVDR Art 79
- MDR Art. 2 (64), IVDR Art. 2 (67)
- MDR Art. 2 (65), IVDR Art. 2 (68)
- MDR Art. 85, IVDR Art. 80
- MDR Art. 86, IVDR Art. 81
- A horizontal guidance on the topic of Vigilance is under development and will be provided on:
- MDR Art. 87, IVDR Art. 82
- MDR Art. 88, IVDR Art. 83
- MDR Article 92 and IVDR Article 95
- Electronic templates for Trend Reporting are under development and will be provided here:
- Use of IMDRF codes is desirable also in the context of periodic post-market surveillance reports and periodic safety update reports as referred to in Articles 85 and 86 of the MDR.
- Directive (EU) 2016/1148 of the European Parliament and of the Council of 6 July 2016 concerning measures for a high common level of security of network and information systems across the Union,
- Regulation (EU) 2016/679 of the European Parliament and of the Council of 27 April 2016 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data, and repealing Directive 95/46/EC (General Data Protection Regulation) (Text with EEA relevance),
- ANSM: Cybersecurity of medical devices integrating software during their life cycle; currently under development 34 Cyber Security Requirements for Network-Connected Medical Devices; see
- eHealth Suisse: Guideline for app developers, manufacturers and distributors; see
- Regulation (EU) 2019/881 of the European Parliament and of the Council of 17 April 2019 on ENISA (the European Union Agency for Cybersecurity) and on information and communications technology cybersecurity certification and repealing Regulation (EU) No 526/2013 (Cybersecurity Act) (Text with EEA relevance)
- Medical Device Cybersecurity Guide; see: