MDCG 2025-9:突破性医疗器械(BtX)指南
文件编号:MDCG 2025-9
发布日期:2025年12月
适用法规:EU MDR 2017/745 / EU IVDR 2017/746
官方PDF:下载
注意:BtX试点计划预计于2026年Q2启动。
文件目的
本指南介绍了欧盟医疗器械"突破性器械"(Breakthrough Device, BtX)路径,为满足条件的创新器械提供加速合规评估通道。
BtX路径概述
背景
BtX路径是欧盟委员会为促进创新医疗器械快速进入市场而建立的机制,类似于美国FDA的"突破性器械"指定(Breakthrough Device Designation)。
适用范围
BtX路径适用于同时满足以下条件的器械:
- 创新性:代表重大技术突破,或与现有治疗方案相比具有显著优势
- 严重疾病:用于治疗、诊断或预防严重或危及生命的疾病
- 未满足需求:针对目前缺乏有效治疗方案的疾病
BtX路径的优势
| 优势 | 说明 |
|---|---|
| 加速审查 | 公告机构优先处理BtX申请 |
| 早期互动 | 与公告机构的早期科学建议 |
| 滚动审查 | 可在完整技术文件提交前开始审查 |
| 专家支持 | 可获得MDCG专家小组的意见 |
申请条件
资质要求
申请BtX指定须提供:
- 器械描述和创新性说明
- 目标疾病的严重性证明
- 与现有治疗方案的比较
- 初步临床证据(如有)
申请流程
制造商提交BtX申请
↓
公告机构初步评估(30天)
↓
BtX指定决定
↓
早期科学建议会议
↓
滚动技术文件审查
↓
最终合规评估
↓
CE标志颁发对临床证据的影响
BtX路径不降低临床证据要求,但允许:
- 在临床数据不完整时启动审查
- 基于中期数据颁发有条件CE标志
- 要求上市后临床研究作为条件
与美国FDA BDD的比较
| 方面 | EU BtX | FDA BDD |
|---|---|---|
| 法规基础 | MDR/IVDR | 21st Century Cures Act |
| 适用范围 | MDR + IVDR | 器械 + IVD |
| 加速机制 | 优先审查 + 早期互动 | 优先审查 + 互动 |
| 上市后要求 | 可能有条件 | 可能有条件 |
相关文件
官方文件全文
MDCG 2025-9
Guidance on Breakthrough Devices (BtX) under Regulations 2017/745 & 2017/746
December 2025
This document has been endorsed by the Medical Device Coordination Group (MDCG) established by Article 103 of Regulation (EU) 2017/745. The MDCG is composed of representatives of all Member States and it is chaired by a representative of the European Commission. The document is not a European Commission document and it cannot be regarded as reflecting the official position of the European Commission. Any views expressed in this document are not legally binding and only the Court of Justice of the European Union can give binding interpretations of Union law.
1. Abbreviations and Terminology
For the purpose of this guidance document, the following terms are defined as follows:
- BtIVD - Breakthrough IVD - an in vitro diagnostic medical device that qualifies as breakthrough per section 4 of this guidance
- BtMD - Breakthrough medical device - a medical device, not being an in vitro diagnostic medical device, that qualifies as breakthrough per section 4 of this guidance
- BtX - Breakthrough device - collective term for breakthrough medical devices and breakthrough IVDs
- CECP - Clinical evaluation consultation procedure
- CER - Clinical evaluation report, as referenced in MDR Annex XIV Part A
- CI - Clinical investigation of a medical device, as defined in MDR Article 2(45)
- Device - collective term for MD and IVD
- GSPR - General safety and performance requirements, referring to the requirements in Annex I of the MDR and IVDR as applicable
- IVD - an in vitro diagnostic medical device, according to IVDR Article 2(2)
- IVDR - Regulation (EU) 2017/746 on in vitro diagnostic medical devices
- MD - a medical device, other than an IVD, as defined in MDR Article 2(1)
- MDCG - Medical Device Coordination Group
- MDR - Regulation (EU) 2017/745 on medical devices
- NB - Notified body
- NCA - National competent authority
- PER - Performance evaluation report, as reference in IVDR Annex XIII Part A
- PMCF - Post-market clinical follow up, as referenced in MDR Annex XIV Part B
- PMPF - Post-market performance follow up, as referenced in IVDR Annex XIII
- PMS - Post market surveillance
- PS - Performance study of an IVD, as defined in IVDR Article 2(42)
- RCT - Randomised Controlled Trial
- SOTA - State Of The Art
- SME - Micro, small, and medium enterprises, as described in Recommendation 2003/361/EC
- SSCP - Summary of safety and clinical performance
- SSP - Summary of safety and performance
2. Introduction
In certain circumstances, a medical device (MD) or in vitro diagnostic medical device (IVD) may be described as a breakthrough device (BtX) when it represents an innovation that is expected, based on available information, to offer a novel diagnostic or therapeutic option addressing an unmet medical need for a particular patient population with serious and lifethreatening diseases or conditions when compared with state of the art.
In alignment with the MDR and IVDR, this guidance presents recommendations for facilitating a streamlined pathway for CE marking and market access of breakthrough devices, to allow for timely patient access without compromising the clinical evidence requirements for these devices. It describes the opportunities and benefits available to manufacturers of BtX with the aim of eliminating undue delays and supporting the safe innovation and availability on the market of BtX. This document provides guidance for a process that accommodates the needs of these devices and provides considerations for the clinical or performance evaluation of breakthrough medical devices (BtMD) and breakthrough in vitro diagnostic medical devices (BtIVD) .
In the absence of specific provisions in the MDR or IVDR, the application of regulatory requirements to breakthrough devices should be balanced and proportionate in light of Article 35 of the Charter of Fundamental Rights (health care) 1 , so that the pre-market clinical evidence requirements are sufficiently met without unduly hindering or delaying patient access to these breakthrough devices. As such, clinical evidence requirements should be balanced between pre-market and post-market to ensure patient safety and clinical benefit while fostering innovation. To that end, this document provides guidance with the aim of facilitating the appropriate, timely, and proportionate pre-market collection of clinical data for MDs and data on the performance for IVDs. In addition, guidance is provided with regard to the collection of PMS and PMCF/PMPF data for breakthrough devices.
This guidance has been developed so that a MD or IVD can be designated as a BtMD or BtIVD at an early stage of its development, months to years before it is expected to achieve market access. BtX designations may be granted to multiple MDs or IVDs in development that have the same proposed intended purpose(s) or indication(s), and a BtX's breakthrough status is not influenced by the placement on the market of another MD or IVD with the same intended purpose. Once designated, a BtX can avail of certain supports and benefits to aid in its development and timely market access, as described later. A device's breakthrough status may remain valid for as long as needed for the purposes of applying this guidance.
It is important to note that the status as a breakthrough device does not confer market exclusivity for that device. The criteria should not be interpreted so as to prevent more than one device in a given therapeutic area being designated as a BtX. Similarly, the existence of a BtX in a specific therapeutic area is not alone a reason to prevent a manufacturer from justifying BtX status for another similar device intended for use in the same disease or condition.
Of note, in some cases, a BtX might also qualify as an orphan medical device in accordance with MDCG 2024-10. In such cases, the guidance provided in both MDCG 2024-10 and this guidance document may be relevant to the device, and both guidance documents can be considered to complement each other.
3. Scope
This document provides guidance for manufacturers, expert panels and notified bodies on the process and regulatory considerations relevant for qualifying, assessing and certifying breakthrough medical devices and breakthrough IVDs. It describes some key roles of actors in this context, including EMA expert panels and national competent authorities, and outlines the supports and opportunities available to manufacturers of BtX. This guidance also provides considerations on the clinical evaluation / performance evaluation of BtX, as well as the role of non-clinical data and preclinical evaluation, and post-market clinical follow-up / performance follow-up for these devices.
This guidance document may apply to medical devices and IVDs across all technologies and risk classifications.
Custom-made devices , in-house devices , and products listed in MDR Annex XVI without an intended medical purpose are outside the scope of this guidance.
4. Breakthrough device criteria
4.1 Criteria
For the purposes of this guidance, a MD or IVD will be considered a breakthrough device if it meets each of the following criteria:
1. Novelty
The device introduces a high degree of novelty with respect to the device technology, the related clinical procedure, and/or the application of the device in clinical practice,
AND
2. Positive clinical impact
The device is expected to provide a significant positive clinical impact on patients or public health, for a life-threatening or irreversibly debilitating disease or condition, by either of the following:
- Offering a significant positive clinical impact on patients or public health compared to available alternatives and the state of the art, OR
- Fulfilling an unmet medical need where there is an absence or insufficiency of available alternative options for that purpose.
4.2 Considerations on meeting the criteria
This section should be read in conjunction with the criteria outlined in Section 4.1 above, which define the conditions under which a medical device or in vitro diagnostic medical device (IVD) may be considered a Breakthrough Devices (BtX) for the purposes of this guidance.
The intention of this section is to provide further considerations and interpretative guidance to support the assessment of whether a device meets each of the BtX criteria - namely, novelty and positive clinical impact. These considerations aim to promote consistency and transparency in the application of the criteria while allowing flexibility to accommodate the diversity of technologies, clinical contexts, and intended purposes falling within the scope of the Regulations.
The examples and elements described in the following subsections are illustrative rather than exhaustive. They are intended to guide manufacturers, notified bodies, and competent authorities in determining whether a device demonstrates a sufficient degree of novelty and potential for significant positive clinical impact to be regarded as a breakthrough technology.
4.2.1 Degree of novelty
Novelty is mentioned directly in both the MDR and IVDR. In general, novelty relates to certain characteristics of the device, the related clinical procedure, or the application of the device in clinical practice, that otherwise do not have an established history of use in the state of the art 2 . A high degree of novelty is required for a device to be considered BtX;
Novelty with respect to device technology may include novelty of the:
- materials, including their composition, chemical, physical and biological characteristics, duration of contact of materials with human tissues or body fluids, or changes in the release characteristics of substances,
- design, including new or modified specifications and properties,
- manufacturing process,
- sustainability, circularity, durability, reusability or environmental impact of materials, design or manufacturing process,
- mechanism of action,
- incorporated technologies or components that are integral to the functioning of the device, e.g., biomarkers, analytes, test platform, or combination/integration with other devices,
- type of sample or specimen,
- device portability e.g. near patient tests/self-tests,
- level of automation e.g. laboratory workflow automation,
- site of application for an established material, leading to new/modified contact with and/or mechanical loading of the same or different tissues,
- technologies including medical device artificial intelligence (MDAI), devices with nanotechnology or advanced materials, or devices for precision medicine.
Novelty with respect to the related clinical procedure or to the application of a device in clinical practice 3 may include novelty of the:
- intended purpose or indication, for example for IVDs detection/measurement of biomarkers to a different/new clinical condition
- intended user, e.g., level of public access, application of the device to lay users,
- mode of application, e.g., novel procedural approach or deployment methods, early detection/prediction/screening, monitoring/disease management,
- or application of existing technologies in a novel context,
- interface or interaction of patients or users with the device, including control, maintenance, and adjustment.
Additional considerations
Where a device's novelty constitutes a e.g. first-in-class device, the first certification of that type of device 4 , or is introducing an innovation that is expected to result in a paradigm shift, that device is more likely to represent sufficient novelty with respect to qualifying as breakthrough.
Unless the device offers a substantial and clinically meaningful deviation from the relevant state of the art, it may be challenging for the device to qualify as breakthrough the novelty is limited to incremental, sustaining improvement or iterative changes.
As novelty increases, uncertainty may also increase regarding the expected safety or performance of the device in question. This reflects the fact that devices with a high degree of novelty are divergent from alternatives and state of the art, and thus there is less relevant supporting information from similar devices to help inform the risk evaluation for the novel device. Where a high degree of novelty is associated with such increased uncertainty, the device may only be considered BtX if there is adequate justification for how the device is expected to provide a significant positive clinical impact, i.e., for how it is expected to contribute to a clinically meaningful improvement in patient or public health outcomes compared to available alternatives and the state of the art.
This is further illustrated in Table 1, see Appendix.
4.2.2 Significant positive clinical impact on patient or public health
A MD or IVD can be considered breakthrough only if it is reasonably expected to provide a significant positive clinical impact to patient or public health for life-threatening or irreversibly debilitating diseases or conditions, in the context of current alternative options (if any) and state of the art. The device's expected clinical impact needs to be compared with alternatives and the state of the art in terms of clinical performance, clinical benefit, safety, risks, and quality of life for patients. Where there are no or insufficient alternatives available, there needs to be sufficient justification as to how the device in question addresses an unmet medical need, on an individual and/or population level, that is not fulfilled by the current state of the art.
The manufacturer should justify how the expected clinical impact of the device can contribute to a clinically meaningful improvement in health outcomes on an individual (patient health) and/or population (public health) level 5 . When assessing the clinical impact of a device, the totality of expected benefits and foreseeable risks need to considered and sufficient rationale is needed to justify how the expected clinical impact will be positive. The following aspects should be considered when assessing the clinical impact of the device:
Clinical impact on patient health: effects on an individual level
Clinical impact on patient health is understood, in this context, as the totality of benefits, harms and related risks at the individual level. Clinical impact on patient health refers to a MD or IVD's expected or potential impact on:
- diagnosis, treatment, management, or prevention of a life-threatening or irreversibly debilitating disease or condition,
- clinical outcomes, for example differences in safety and/or clinical performance compared with state of the art and alternatives (if any) leading to changes in: mortality, morbidity, health-related quality of life, burden of treatment, frequency or duration of hospitalisations, mode of administration, severity, duration and frequency of effects, need for medical or surgical re-intervention, etc.
- provision of care to individuals or specific groups of patients, e.g.: differences in mode of administration, procedural efficiency, or cost-effectiveness, resulting in clinical benefits or advantages to patients compared with state of the art,
- risks, hazards, and negative clinical outcomes, related to the use of the MD or IVD,
- risks related to incompatibility with the use of other MDs or IVDs,
- risks related to specific groups of patients, with a focus on vulnerable populations,
- risks related to medical device dysfunction due to reasonably foreseeable misuse.
Clinical impact on public health: effects on a population level
Clinical impact on public health is understood as the net potential benefits and risks stemming from the clinical impacts on a population level. Clinical impact on public health refers to a MD or IVD's expected or potential impact on:
- individual patient health, cumulatively expressed on a population level,
- prevention, diagnosis, management, and treatment of serious public health threats 6 and conditions associated with significant risk to public health,
- response to and management of diseases and conditions that represent local, regional, national, European, and/or international health emergencies,
- system-level benefits, for example, by enabling clinically accepted treatment pathways to be delivered in a significantly more cost-effective, efficient, scalable, and/or simplified manner with a greater capacity compared with state of the art,
- market penetration, which may lead to an increased uptake and exposure of the device to patients, which may in turn increase the probability of benefits and/or harms on a population level.
For the purpose of meeting the BtX clinical criterion, a complete set of clinical data is not required at the time of designation. Instead, the manufacturer should demonstrate a reasonable expectation that the device could provide for more effective treatment or diagnosis of the disease or condition identified in the proposed indications for use and intended purpose. This includes a reasonable expectation that the device could perform as intended and that it could more effectively treat, diagnose, prevent, or improve the management of the relevant disease or condition, compared to available alternatives and the state of the art. To demonstrate meeting the BtX clinical criteria, manufacturers could include relevant information from scientific literature, data supporting scientific validity, pre-clinical data (e.g., bench, animal, simulation), preliminary clinical data and performance data.
The level and type of information needed may vary depending on the intended use of the device, its technology and features, and the available standard of care alternatives. The manufacturer should consider the totality of information regarding the proposed device, its function, potential for technical success, potential for clinical success, potential for a clinically meaningful impact, and its potential benefits and risks. For new devices, information relevant for estimating the clinical impact will typically come from the clinical evaluation (for MD) or performance evaluation (for IVD) during the pre-market phase. For modifications of existing devices, evaluation of the clinical impact may also draw on relevant post-market information.
Information from medical literature (e.g. systematic reviews, clinical treatment guidelines, consensus statements from relevant scientific and clinical experts or medical societies) may be used to support the justification of the expected clinical impact. For example, the literature may highlight relevant gaps in clinical management of the disease in the existing state of the art, which can help justify how the proposed breakthrough device is needed to address those gaps. Relevant non-clinical, clinical, and performance data on the device, available alternatives (if any), and the state of the art may be used to support a statement that the breakthrough device will provide an expected positive clinical impact.
Examples that may offer positive clinical impact on patient or public health, compared to alternatives, include:
- a device intended to treat, prevent, or diagnose a life-threatening or irreversibly debilitating disease or condition that results in clinically significant improvements in patient-relevant clinical outcomes,
- an IVD, including companion diagnostics, intended to provide information for a lifethreatening or irreversibly debilitating disease or condition in a way that could lead to clinically significant improvements in clinical outcomes (e.g., earlier diagnoses, greater sensitivity and specificity, more effective treatment),
- a device intended to treat or prevent a severe side effect associated with the treatment of a life-threatening or irreversibly debilitating disease or condition,
- a device intended to treat a life-threatening or irreversibly debilitating condition that has a significantly lower risk of harm compared to alternatives.
- an IVD intended to rapidly diagnose or detect a disease with potential to cause a public health emergency, that is significantly more cost-effective, efficient, and scalable, with a greater capacity and speed compared.
- a device intended to facilitate the delivery of early and effective care to patients in the community in a more cost-effective manner, leading to clinical benefit to the individual patient and system-level benefits through reductions in hospitalisations and capacityrelated burdens on the healthcare institution.
4.2.3 Life-threatening or irreversibly debilitating disease or condition
In this context, a disease or condition is considered life-threatening if it is highly likely to result in death in the absence of major medical intervention. Treatment options may be limited in such cases, and typically major medical intervention is required to interrupt the course of the disease or condition 7 .
A disease or condition is considered irreversibly debilitating if it is associated with morbidity that has substantial impact on day-to-day functioning or quality of life for an individual, population or subpopulation. Short-lived or self-limiting morbidity is generally not considered irreversibly debilitating; however, in certain cases, diseases or conditions that are persistent or recurrent may be considered irreversibly debilitating.
Determining if a disease or condition is irreversibly debilitating should be based on its impact on factors such as survival, activities of daily living, and the likelihood that there will be disease progression and increase in severity over time if left untreated or under the current state of the art of clinical practice. Examples of such diseases or conditions may include cancer, neurodegenerative diseases, diabetes mellitus, stroke, and ST-segment elevation myocardial infarction.
4.2.4 Available alternatives and the state of the art
Manufacturers are required to describe the current state of the art for management of the disease or condition in question, including determining whether relevant available alternatives exist on the EU market (i.e., whether there is an alternative drug, biological product, MD/IVD, combination product, or clinical procedure available on the EU market for the same specific indication(s)).
There may be a number of currently available potential alternatives with varying relevance in the diagnosis, prevention, or treatment of a life-threatening or irreversibly debilitating disease in the EU, including options that are no longer used or are used only rarely. Therefore, when considering potential alternatives, the manufacturer should focus on options that reflect the current state of the art for the same specific indication(s), including the disease stage, for which their device is being developed.
In evaluating the state of the art, consideration should be given to any relevant scientific standards and guidance, including recommendations by appropriate authoritative scientific bodies, clinical practice guidelines, and other reliable information that reflects current clinical practice. When a proposed BtX is indicated for a subgroup within a broader disease population, the state of the art for the broader population should be considered available alternatives for the subgroup, unless duly justified.
PART A: Pre-clinical, Clinical, and Performance Evaluation Considerations
5. General considerations
For BtX, as for all devices, clinical evidence 8 must demonstrate compliance with the requirements of the MDR or IVDR, including that the benefit-risk ratio is acceptable. To that end, the manufacturer is required to conduct a clinical evaluation (for MDs) or a performance evaluation (for IVDs). 9
As described in section 4.2.2, a device with a high degree of novelty may be associated with uncertainty regarding the device's expected safety or performance, due to limited prior scientific knowledge or data from similar devices (if any). These devices may be associated with new risks or unexpected side effects; the necessary clinical evidence has to appropriately address this uncertainty.
For BtX, a balanced approach is needed to ensure that the level of uncertainty associated with the device doesn't unnecessarily prevent timely patient access to devices that provide significant positive clinical impact compared with the state of the art and alternatives (if any). When comparing the life-cycle clinical evidence requirements of BtX with other devices, the overall clinical evidence necessary does not differ. However, for BtX, given the potential for significant positive clinical impact, consideration should be given to whether the necessary longer term data 10 can be collected in a way that would allow for more timely availability of these devices to patients without compromising the clinical evidence needed to confirm the acceptability of the benefit risk ratio.
In particular in cases where the BtX is fulfilling unmet medical needs it may be acceptable for a BtX to be placed on the market with a higher level of uncertainty and more limited pre-market clinical evidence provided that a well-defined plan is in place to collect confirmatory data through comprehensive PMS and PMCF/PMPF.
In summary, provided that the available pre-market data give an appropriate level of assurance of safety and clinical performance of the device and support the manufacturer's claim of significant positive clinical impact compared with the state of the art and available alternatives, it may be appropriate for certain confirmatory data to be collected through welldefined, scientifically valid, and milestone-based PMS, PMCF/PMPF plans (e.g. studies, investigations, other activities).
With respect to clinical evidence requirements, it may, in general, be acceptable to place a BtX on the market provided that all of the following can be justified for the device in question:
- the available data 11 sufficiently demonstrates:
- that the benefit-risk ratio is acceptable; and
- that it is reasonably expected the device will provide a clinical benefit 12 , taking into account the clinical condition, the state of the art, and the safety of patients,
- the manufacturer has an appropriate PMS, PMCF/PMPF plan (see section 9), with a clear justification as to why the clinical data to be collected through PMCF/PMPF are not being collected in the pre-market phase,
- users and patients will be adequately informed (e.g. by provision of information in the labelling, IFU, SSCP/SSP (if applicable), and/or other accompanying documentation) of the breakthrough status of the device and instructions to users on how to report incidents, complaints, and other clinical experience to the manufacturer,
- appropriate measures are in place to meet any conditions or provisions of certification that have been imposed by a notified body.
Risk management
It is good practice in the planning phase of the device development to document in the risk management file how evidence will be provided to demonstrate that risks, including in particular increased risks or new risks arising from the innovative nature of the design or technology, have been identified and effectively mitigated. Risk management is a lifecycle process, which must be linked also in an early stage of the development with the clinical evaluation process to ensure that the residual risks have been weighed against the benefits offered by the BtX device. This ensures a comprehensive evaluation of safety and performance, while supporting the availability of the innovation's benefit for the patients, as well as during the clinical investigations / performance studies in the treatment group, and during the post-market phase.
In the early stages of developing BtX devices, safety and performance data from patient use are in principle unavailable. This means that the occurrence rates of side effects, incidents, and harms from residual risks may not be fully quantifiable or quantified under certain assumptions. As such, the manufacturer's criteria for accepting risks, when the probability of harms cannot be fully estimated or estimated accurately, become particularly significant. Input from the state of the art assessment will need to provide the risks levels of the currently accepted clinical practice and specific focus on the new design features and/or resulting clinical procedure needs ensure that acceptable risk levels are determined from a safety perspective.
Appropriate technical and clinical inputs should be considered to verify and validate (as far as possible), the assumptions made in relation to the estimated occurrence levels, the predicted harm(s) associated with the hazards, the severity of those harms and the acceptance criteria, all in comparison with current state of the art. An estimation of the occurrence rate is necessary before the clinical investigation(s) / clinical performance study, in order to establish the acceptance criterion which will enable identification of unacceptable risks while the studies are conducted according to the plan and clinical data are gradually collected. These data, including PMCF/PMPF, enable the continuous verification of the initial estimation, and a databased quantification of all known clinical risks and is expected to be incorporated into the risk management process.
Considerations relevant for IVDs
The principles outlined above remain relevant for IVDs, however consideration of IVD specific aspects is warranted.
When it comes to IVD BtX, the performance evaluation should include the evaluation of the analytical performance, scientific validity, and clinical performance. In line with the guidance in the rest of this document, manufacturers should utilise avenues at their disposal to generate data where required and possible.
Similar to BtXMDs, there may be insufficient existing evidence to draw on for certain BtIVDs. In such circumstances it may be challenging to demonstrate scientific validity based on available information. There may be a need to conduct proof of concept studies or other activities to generate evidence to support the scientific validity. Consideration may be given to additional methodologies, e.g. in-silico, to support the evidence body for scientific validity.
Given the potential for limitations with respect to both scientific validity and clinical performance, the robustness of the analytical performance data is key for BtIVDs. The use of novel markers or markers without certified reference materials can lead to challenges in assessing BtIVDs. Where there are no comparative methods, comparisons to other welldocumented methods or the composite reference standard can be used where appropriate and justified (IVDR Annex XIII, 1.2.2). In the absence of comparator assays, consider other means to verify performance e.g. robust orthogonal testing, creation of internal reference material etc.
Considerations relevant for MDAI
In the case of BtX that are or contain one or more high-risk AI systems (MDAI), additional considerations are required due to the simultaneous and complementary application of both the MDR/IVDR and the AI Act. 13 These considerations apply across the entire device life cycle, including design, pre-clinical, clinical, and performance evaluation, post-market surveillance and change management activities.
Particular attention should be given to the quality and representativeness of data sets used for training, validation and testing of MDAI. Manufacturers should implement robust data governance and management practices, ensuring that data are relevant, accurate, complete and representative to the best extent possible, and that appropriate measures are in place to identify and mitigate bias in the data sets used for training, validation and testing, in accordance with the requirements of the AI Act . These practices form a core part of both the risk management system and the clinical evaluation or performance evaluation process, given the significant influence that data integrity has on device safety and performance.
In this context, manufacturers are expected to ensure that the clinical evidence adequately demonstrates the performance, reliability, and generalisability of the MDAI within its intended purpose and intended use environment. The evaluation should take into account the dynamic characteristics of MDAI, including the impact of training, updates, and data drift, as well as the controls established through Pre-Determined Change Control Plans (PCCPs) to manage any modifications in a predictable and transparent manner.
When applying the principles outlined in the IMDRF guidance 'Good Machine Learning Practice for Medical Device Development: Guiding Principles', manufacturers should ensure that the AI system's design and validation processes are consistent with the MDR/IVDR requirements on risk management, clinical evidence, and post-market follow-up. This integrated approach supports a continuous demonstration of conformity, ensuring that the safety and performance of the MDAI remain consistent throughout its lifecycle while preserving the benefits of innovation for patients and healthcare systems.
6. Considerations on non-clinical data and preclinical evaluation
Pre-clinical testing, verification and validation including usability testing are essential to demonstrate the safety of the device and to support a hypothesis that the device is safe and performs as intended. The non-clinical data must sufficiently demonstrate the safety of the device prior to conducting clinical investigations or performance studies. At the time of conformity assessment, clinical/performance data for BtX devices may be limited due to their innovative nature. Non-clinical data should reduce the risk of remaining uncertainties as far as possible and consequentially, this data should be as robust and comprehensive as possible.
For non-BtX devices, manufacturers utilise e.g. existing scientific and clinical literature, validated test methods, product-specific standards, and commonly accepted criteria, which create a basis for demonstrating how the device will perform in the clinical setting and whether it is safe when used as intended. Due to the innovative nature of BtX and the inherent limited experience, it is recognised that there might be specific challenges for pre-clinical evaluations. These include limited appropriate standards for testing, verification and validation, and the lack of availability of reference materials for biocompatibility or appropriate test criteria. The following sections outline some important considerations in the generation of pre-clinical data for BtX devices considering the challenges associated with such devices.
Pre-clinical literature research
Pre-clinical literature search in relation to the materials used, the technologies applied, specific design elements, etc. should be conducted and the results analysed to:
- establish the current state of the art in relation to any alternative therapies, if available,
- identify risks associated with the materials, technologies used etc in the subject device,
- support the justification of superiority/breakthrough of the design or technology of the subject device in achieving its intended safety or performance characteristics,
- establish that there are sound scientific principles supporting the use of the novel device's aspects in question
Such data, even if originating from fields/areas unrelated to medical applications, should be considered and analysed if relevant to the intended purpose or otherwise justified, and demonstrating safety and performance of the device.
Biological safety evaluation
The biological safety evaluation for BtX devices should be conducted following the same process as for non-BtX devices, within the context of the risk management process. Any specific risks introduced by novel materials or manufacturing technologies/methods in relation to the biological safety of the device must be carefully evaluated considering the intended use (including all intended users and target patient groups, medical indication, part of the body or type of tissue interacted with, duration of contact, etc). Together with the information on the intended use and any reasonably foreseeable misuse, available information on chemical, physical and biological characteristics forms an important starting point. Depending to what extent these data are sufficient to address identified biological risks, an appropriately justified strategy and planned content for the biological evaluation of the medical device, including any requirements for additional data generation must be developed, as well as the relevant endpoints to be evaluated.
For novel medical devices, depending on their characteristics it can be appropriate to evaluate additional biological effects other than those generally indicated for evaluation. Potentially, relevant endpoints include e.g. cytotoxicity, irritation/sensitisation, systemic toxicity, local effects after tissue contact, genotoxicity, carcinogenicity, haemocompatibility, developmental/reproductive toxicity, immunotoxicity and neurotoxicity. Important elements include for example, toxicologic evaluation of leachables and contaminants, degradation characteristics and degradation products in the case of absorbable devices, specific biological risks in case of nanomaterials. In certain cases, toxicokinetic studies to determine absorption, distribution, metabolism and excretion may be required.
Animal testing should be carried out only when other available scientific data and in vitro studies cannot provide sufficient information to address the safety of a medical device. If animal testing is required, it should be performed according to the relevant standards and with appropriate study designs. The 3Rs principle, replacement, reduction, and refinement should be applied, in order to optimise animal welfare.
Bench testing
For BtX devices, rigorous bench testing including relevant verifications and validations, is most likely necessary for addressing technological uncertainties, especially in the absence of applicable standards. If relevant standards exist, these should be utilised where possible in bench testing. In the absence of standards, manufacturers are required to define critical performance parameters and relevant acceptance criteria based on clinical simulation, analogous methodologies, or theoretical models ('in silico' - see below). This involves developing detailed, custom test protocols for various conditions, including worst-case scenarios. Any test methodology chosen should be validated and appropriate for providing evidence towards the intended purpose of the device, The choice of the model(s) should be justified considering the intended use and the patient population.
Bench testing results from early prototype evaluation provide the evidence that risk mitigation actions are effective and as such feed into risk management, allowing demonstration appropriate risk control before a device is applied in a patient setting.
In silico modelling & simulation
The design and pre-clinical development of BtX, including the rationalisation of animal testing, can be supported and accelerated through the use of digital models and simulations. These simulations and digital models can provide valuable input for establishing and updating the Clinical Evaluation Plan or Performance Evaluation Plan, and can complement the pre- market clinical or performance data. It should be noted that any in silico model intended to support the assessment of BtX is required to demonstrate its credibility through appropriate verification and validation activities prior to being accepted as part of the evidence base in support of the assessment of BtX devices.
Long-term non-clinical data
As a general principle, the MDR and IVDR require that safety and performance of the device is maintained during the shelf life and time the device remains in the patient or in use (lifetime). Due to the innovative nature of BtX devices, some real time non-clinical data may not be available at the time of the clinical investigation, performance study or certification. In such
Medical Device Coordination Group Document cases, simulated use/ageing data have to be considered as preliminary evidence.
For long term performance data, e.g. the resistance of implants against cyclic loading, or the long-term stability of electrodes in active implants, simulated use models may serve to generate preliminary data for the device's long-term performance in the patient. Manufacturers are expected to provide a plan for confirming simulated long-term test results by real-time and real-world data. In this context, the manufacturer is required to comprehensively identify any limitations of the simulated use testing data which require support by long-term real-world data. In any such long-term study planning, adequate interim analysis timepoints are expected to be specified, to identify deviations from the initial assumptions on the device performance and safety at an early stage to allow to initiate countermeasures where necessary.
Usability aspects
For BtX devices, robust usability testing is essential for safety and performance, requiring a strong useability engineering process approach from concept to post-market. If the BtX device utilises a technology that hasn't been used before, manufacturers should understand and clearly document the diverse user profiles, use environments, and clinical workflows where the device is expected to be used (incl. reasonably foreseeable misuse). The user interfaces (where applicable) should be intuitive and ergonomically designed to minimize errors or harm to the patients/users. This includes measures such as clear UIs, effective alarms, and comprehensive instructions, all aimed at preventing use-related hazards. Additionally, appropriate training should consider learnability.
Critical to this process are iterative design and extensive usability testing, starting with early "formative" evaluations and culminating in "summative" validation with representative users in realistic scenarios. Manufacturers should also integrate proactive risk management throughout development, ensure standards are duly considered, and implement robust postmarket surveillance to continuously improve device usability and ensure ongoing patient safety.
Novelty in manufacturing aspects - validation of the manufacturing processes including appropriate in-process and final inspections
BtX devices, amongst others, may utilise novel materials, or novel manufacturing technologies not used before in medical devices. It is expected that the technical documentation includes clear and traceable device specifications, together with a description of the manufacturing processes and their validation, the incoming inspection, in-process controls and continuous monitoring and final product testing. In the absence of broadly accepted solutions, the methods and acceptance criteria employed to furnish evidence that the finished device meets the specifications set out by the manufacturer must be supported by a documented scientific justification by the manufacturer. This description enables the competent authority and/or notified body to verify the appropriateness of the process validation during the on-site audit, inspection and/or technical documentation assessment. Also risks related to the manufacturing and use of novel materials must be duly considered as applicable.
7. Considerations on premarket clinical evidence
7.1 Clinical Evaluation Overview
As with all devices, the manufacturer of BtX has to establish and update a clinical evaluation plan (CEP), with particular attention to the following steps:
- a detailed description of intended clinical benefits to patients with relevant and
Medical Device Coordination Group Document specified clinical outcome parameters;
- a specification of methods to be used for examination of qualitative and quantitative aspects of clinical safety with clear reference to the determination of residual risks and side-effects;
- an indicative list and specification of parameters to be used to determine, based on the state of the art in clinical practice, the acceptability of the benefit-risk ratio for the various indications and for the intended purpose or purposes of the device;
- an indication how benefit-risk issues relating to specific components such as use of pharmaceutical, non- viable animal or human tissues, are to be addressed;
- a clinical development plan including premarket clinical investigations and PMCF activities.
As the CEP may be used to guide the provision of scientific advice, this document should clearly identify the innovative nature of the device. The CEP should outline the special challenges relating to balancing the uncertainty relating to the device's innovative nature and the timely lifecycle generation of clinical data. Given the novel nature of BtX, clinical investigations and performance studies are considered a key source of pre-market clinical evidence for the clinical evaluation.
7.2 Clinical Investigations / Performance Studies
BtMD that are implantable devices and/or class III devices require the performance of clinical investigations in accordance with MDR Article 61(4). Similarly, for BtIVD, clinical performance studies in accordance with Section 2 of Part A of Annex XIII must be carried out unless it is duly justified to rely on other sources of clinical performance data 14 .
When designing clinical investigations or performance studies for BtX, the accumulation of clinical data throughout the lifecycle of the device should be taken into consideration. It should be considered that premarket studies may focus on safety and short- and medium-term performance to attain sufficient clinical data which enables timely patient access to innovative devices which can produce significant positive clinical impact or fulfill an unmet medical need.
In the absence of traditional approaches to generating clinical evidence for BtIVDs, such as comparison to other well-documented methods or reference standards, a clinical performance study comparing performance of the novel device to the current clinical standard practice is required. Nevertheless, there may be challenges in generating clinical performance data in a timely fashion. The use of Bayesian methodologies to support generating clinical performance data may be warranted. In addition, simulations may be considered to supplementing clinical performance data from in-vitro testing provided care is taken to avoid overinterpretation and the limitations are clearly documented.
As such, while short- and medium-term performance data may be accepted at pre-market stages, the data should be supplemented by post market clinical investigations, other PMCF or PMPF activities.
Appendix A.2 provides further considerations on clinical investigations for breakthrough devices, with particular attention to:
Study design
Objectives
Endpoints
Choice of comparator/control
8. Post market surveillance and PMCF/PMPF
Implementing a comprehensive PMS system is essential for monitoring the continued safety and performance for BtX. 15
The clinical data collected from PMS, including serious incidents, should be carefully evaluated and acted upon as part of the life-cycle evaluation of these devices. In addition to the need for collecting any missing supportive data and evidence as part of the post-market stage, it is foreseeable that some potential rare or latent risks may not be fully characterised at the time of conformity assessment; in this context, it may be appropriate to further characterise these risks or long-time performance through appropriate and scientifically robust PMCF/PMPF.
When it comes to BtIVD, an increased focus of PMPF should be on confirming the performance characteristics. This may include confirmation of a combination of scientific validity, analytical performance, and/or clinical performance through proper post market Performance Follow-up activities. The focus should be on generating data to reduce uncertainties. This may involve PMPF studies, use of External Quality Assurance schemes, monitoring of real-world performance of the IVD or other activities.
Where necessary, the manufacturer should aim to prospectively enrol a representative majority of patients into pre-defined and structured PMCF/PMPF activities with appropriate length of follow-up, until such a time as the data is sufficient for confirming the safety and performance throughout the expected lifetime of the device.
As discussed in PART B, it may be appropriate for specific conditions or provisions of certification to be defined by the notified body for a BtX. These conditions may include the completion of specific PMCF/PMPF activities with pre-defined milestones and timelines for completion of these activities. 16
8.1 PMCF / PMPF Plan
It is essential that there is an appropriate and comprehensive PMCF/PMPF plan that aims to further confirm the BtX's safety and performance throughout its expected lifetime, confirm the continued acceptability of identified risks, and detect emerging risks on the basis of factual evidence. It should be ensured and verified that the PMCF/PMPF plan is implemented and followed to completion. This plan should outline how risks will be monitored, assessed, and mitigated throughout the PMCF/PMPF, and outline how the manufacturer will take appropriate action where these data raise new concerns regarding the safety or performance of these devices. Documents related to specific planned PMCF activities may be provided at the time of conformity assessment, for example Clinical Investigation Plans for planned PMCF investigations.
Due consideration needs to be given with respect to the potential challenges that may be faced during execution of the PMCF/PMPF plan, which may require adjustment of the expected time frame and milestones. Continued discussion between the manufacturer and the notified body
Medical Device Coordination Group Document may be appropriate to discuss any challenges or substantial changes faced with respect to delivery of the PMCF/PMPF plan.
Please refer to MDCG 2020-7 and MDCG 2020-8 for general guidance on PMCF plans and PMCF evaluation reports respectively.
8.2 PMCF investigations / PMPF studies
With respect to the general requirements and expectations for PMCF investigations / PMPF studies, particular attention should be made in the following areas for BtX:
- Consider any possible condition(s)/specific provision(s) provided by the notified body in the certification and their impact on the PMCF investigation / PMPF study.
- Consider any rare or latent risks that may not be fully characterised at the time of conformity assessment that are intended to be further characterised through appropriate PMCF investigations / PMPF studies.
- PMCF investigations / PMPF studies should allow for continuous monitoring and evaluation of these devices over the expected lifetime of the device. Long-term followup is especially important in diseases or conditions with slow progression or where the benefits or risks of the device may manifest over an extended period.
- Where possible, for the duration of recruitment into a PMCF investigation / PMPF study, the manufacturer should plan to enrol a representative majority 17 of patients/patient samples exposed to the device in each study site. This is particularly important for devices that carry significant risks (i.e. high residual risks or risks of causing serious harm) and should take into consideration the clinical data available at the time of CE certification, especially data that demonstrates device safety.
- PMPF studies should focus on confirming the BtIVD performance which was established in the pre-market phase. In particular this includes seeking to address the uncertainties faced during BtIVD development. For example, where there were challenges in generating scientific validity data a focus of PMPF would be to confirm the scientific validity. Similarly, if limitations in sample size were present an emphasis on the clinical performance assessment from a larger population would be appropriate.
8.3 Registries
In relation to BtX, and in particular for high risk interventional or implantable BtMDs, registries are considered as a valuable tool in building a broad and comprehensive knowledge base that can follow the device through its lifetime. As such, it is recommended that BtMD are enrolled in registries as part of PMCF plan where appropriate and feasible.
The manufacturer should identify and, where appropriate, support the development of suitable registries for collecting representative data on patients with the disease/condition as well on those receiving the specific device, with the aim of comparing outcomes in patients across Europe and worldwide. Where available and suitable, manufacturers are encouraged to use registries established and governed by national bodies or speciality medical associations.
The manufacturer should use available guidance and suitable methodologies to demonstrate sufficient access to, and quality of, the data within the registries, for the purposes of confirming the safety and performance of the device throughout the device's life cycle.
For BtIVDs EQA schemes may provide another source of PMPF data similar to registries for BtMDs.
8.4 Other PMCF/PMPF data
In the post-market setting, PMCF/PMPF data can be collected from sources other than through PMCF / PMPF studies. These sources are often referred to as 'real world data' and can be used to generate 'real world evidence'. This clinical data is collected in the postmarket setting during the routine use of the device in clinical practice. The evaluation of realworld data should aim to detect any rare complications and understand any factors that may affect the clinical performance of the device. For BtIVDs the data obtained through use of real world use may be a useful and practical source of PMPF data.
9. Clinical / Performance Evaluation Report (CER / PER)
The general MDR and IVDR requirements 18 regarding the CER and PER apply to BtMD and BtIVD respectively. Within the CER/PER, the manufacturer of a BtX should ensure that the following information is presented in sufficient detail:
- summary of the state of the art and available alternatives (if any),
- summary of how the device meets the criteria for breakthrough status,
- summary of any relevant interactions, dialogues, or advice from expert panels or notified bodies as described in PART B,
- summary of relevant premarket data including data from clinical investigations / performance studies,
- summary of the PMCF/PMPF plan, including:
- the type of PMCF/PMPF data to be generated to confirm the safety, clinical performance and clinical benefit of the device throughout its expected lifetime,
- information on how this data will be generated in a timely manner,
- justification for collecting this data in the post-market setting rather than premarket,
- confirmation that the manufacturer will prospectively enrol a representative proportion of patients into PMCF/PMPF activities including PMCF/PMPF studies and registries,
- a plan with regular, pre-defined intervals on when the CER/PER will be updated, as well as a plan to update the CER/PER if new information becomes available that may change the benefit-risk profile of the device.
PART B - Procedural Considerations
Expert panels, established under MDR Article 106, are composed of independent scientific and clinical experts and play a key role in ensuring consistent and scientifically sound evaluations of certain high-risk medical devices. The following paragraphs address the designation procedure, as well as different scenarios for the consultation of an expert panel depending on the state of advancement of the device's regulatory status.
10. Involvement of Expert Panels
Expert panels, established under Article 106 of Regulation (EU) 2017/745 (MDR), are composed of independent scientific, technical, and clinical experts and contribute to ensuring that the evaluation of certain medical devices and in vitro diagnostic medical devices (IVDs) is consistent, evidence-based, and scientifically sound. Their involvement enhances regulatory predictability, promotes high public health protection standards, and facilitates innovation in areas of unmet medical need.
This section outlines the potential interactions between the expert panels and manufacturers of devices designated as breakthrough technologies (BtX), including both Breakthrough Medical Devices (BtMDs) and Breakthrough In Vitro Diagnostic Devices (BtIVDs). The engagement of expert panels may occur at different stages of a device's development or conformity assessment process, depending on its maturity and regulatory context. Where necessary, additional scientific or technical experts will be added to the team advising or providing an opinion on specific BtX devices.
For the purposes of this guidance, expert panels may play up to four distinct roles in the BtX framework:
- 10.1. Providing an opinion on BtX status designation;
- 10.2. Providing early scientific advice under Article 61(2) MDR for certain devices;
- 10.3. Providing early pre-clinical or clinical advice, in line with Article 106 MDR; and
- 10.4. Conducting consultations under the Clinical Evaluation Consultation Procedure (CECP), pursuant to Article 54(1) MDR.
The type and extent of expert panel engagement will depend on the stage of device development, the nature of the evidence available, and the regulatory pathway applicable. For example, early-stage devices may benefit primarily from non-clinical or regulatory scientific advice under the pilot framework (section 10.3), whereas more advanced devices subject to notified body review may later enter a CECP process (section 10.4).
In addition to the pre-clinical or clinical advice from the expert panels, after confirmation of the breakthrough status, regulatory advice may also be requested from a competent authority. The EMA expert panel secretariat will act as the contact point for that request and liaise with the relevant authority/ies.
Where relevant, similar principles will apply to BtIVDs, with expert advice tailored to analytical and clinical performance considerations under the IVDR. The Commission, together with the MDCG and expert panel secretariats, will further clarify the modalities for BtIVD interactions to ensure coherence across both legal frameworks.
10.1 Breakthrough status opinion / designation
To be designated as a BtX, manufacturers must request scientific advice from the expert panels established in accordance with MDR Article 106 for an opinion on the BtX status of the device.
An opinion on the BtX status of a medical device or IVD can be requested from the expert panels at any time of development, as long as there is sufficient data supporting the BtX criteria, i.e., an application with detailed supportive evidence on how the device in question meets the criteria set out in section 4 will be required.
The expert panels will endeavour to provide an opinion on the BtX status within 60 days, and to prioritise the applications submitted for BtX status. The Secretariat will notify the MDCG of the expert panels' opinion regarding the breakthrough status of a device as soon as a conclusion is reached. A device which has received such an opinion may be referred to thereafter as BtX designated.
A BtX, once designated, can avail of certain supports and benefits to aid in its development and timely market access. Therefore, the breakthrough criteria have been developed so that they can be applied to a MD or IVD at an early stage of its development. It is possible that BtX designations may be granted to multiple MDs or IVDs in development that have (a) similar proposed intended purpose(s) or indication(s) and that fulfil the breakthrough criteria. It is also possible that in the intervening period between BtX designation and market access, a different MD or IVD may be placed on the market with the same intended purpose. However, once attributed, the breakthrough status of a BtMD or BtIVD may remain valid for as long as needed for the purposes of applying this guidance, and the placement on the market of another MD or IVD with a similar intended purpose does not change the breakthrough status given to a MD or IVD.
10.2 Early advice for BtX class III devices and class IIb active devices intended to administer and/or remove a medicinal product
Manufacturers of class III devices and class IIb active devices intended to administer and/or remove a medicinal product may request early scientific advice in accordance with Article 61(2) of Regulation (EU) 2017/745 (MDR). This provision enables consultation with an expert panel with the aim of reviewing the manufacturer's intended clinical development strategy and proposals for clinical investigation.
Within the context of breakthrough technologies (BtX), this mechanism provides an opportunity for manufacturers to obtain targeted scientific input on their evidence-generation strategy early in the development process and facilitates prioritised assessment by notified bodies (see section 11.1).It is therefore recommended that manufacturers of BtX devices falling within the scope of Article 61(2)use this possibility to seek expert panel advice on their planned clinical development and clinical investigation strategy. Such engagement may also cover proposed post-market clinical follow-up (PMCF) activities where relevant to the demonstration of ongoing safety and performance and in particular if a BtX device is intended to be placed on the market with less reliance on pre-market data compared with similar nonBtX novel devices (see sections 5 and 8).
The expert panel will review the proposed strategy and provide written advice reflecting its scientific and clinical considerations. The expert panels will endeavour to provide their advice within 60 days, and to prioritise the devices that received a positive opinion regarding their BtX status.
The manufacturer must give due consideration to the expert panel's advice - both on the BtX status and on the clinical development approach - and document it in the clinical evaluation report.
To maximise the value of the consultation, requests should clearly identify the specific scientific or methodological questions for which expert input is sought, and highlight how such advice could reduce regulatory uncertainty, support evidence generation, or facilitate timely access to the EU market.
10.3 Early advice for BtX from other device risk classes
In accordance with MDR Article 106(11), for devices from other risk classes than those outlined in section 10.2, the expert panels may be requested to provide advice on the criteria for an appropriate data set for assessment of the conformity of a device, in particular with regard to the clinical data required for clinical evaluation.
For breakthrough in vitro diagnostic devices (BtIVDs), a parallel approach may be envisaged, allowing expert advice from panels established under MDR Article 106. This could cover analytical and clinical performance study design, validation strategies, ensuring alignment with the objectives of the BtX framework.
It is recommended that manufacturers of such BtX devices use this possibility to seek expert panel advice on their planned clinical development and clinical investigation strategy. Such engagement may also cover proposed post-market clinical follow-up (PMCF) activities where relevant to the demonstration of ongoing safety and performance, and in particular if a BtX device is intended to be placed on the market with less reliance on pre-market data compared with similar non-BtX novel devices (see sections 5 and 8).
10.4 Clinical Evaluation Consultation Procedure (CECP)
The consultation of an expert panel in relation to a BtX described in sections 11.2 and 11.3 is independent of the clinical evaluation consultation procedure (CECP) required under MDR Article 54(1). However, the areas that were subject to consultation of the expert panels will be taken into account during the CECP.
The notified body should indicate in their CECP submission if the device has been subject to an advice procedure, and reflect in the clinical evaluation assessment report how the advice has been taken into consideration.
10.5 Advice provided in parallel by the Expert panels and the Member State Coordination Group on Health Technology Assessment (HTA)
Class III implantable devices and class IIb active devices intended to administer and/or remove a medicinal product are eligible for a parallel HTA Coordination Group (HTACG)/Expert Panels (ExP) Joint Scientific Consultation (JSC). Devices that are designated as breakthrough are likely to be exemplary applicants to such procedure.
11. Notified body activities and responsibilities
The notified body's assessment of a designated 19 BtX should take into account the considerations described in this guidance. When performing conformity assessment activities, notified bodies should ensure that the approach applied to BtX devices remains scientifically rigorous, proportionate, and responsive to innovation while maintaining a high level of health and safety protection.
11.1 Prioritisation of BtX and access to structured dialogue
For devices that meet breakthrough criteria, including the potential to address significant unmet medical needs or offer clinically meaningful advantages over existing solutions, notified bodies should:
- Prioritise BtX files during planning and resource allocation, in view of their potential to address unmet medical needs or offer clinically meaningful advantages over existing solutions;
- Engage in early and structured dialogue with manufacturers to clarify expectations regarding the generation of clinical evidence, the design of performance or usability studies, and the development of post-market surveillance and PMCF strategies; and
- Coordinate, where appropriate, with expert panels to ensure coherence between scientific advice and the conformity assessment process, thereby facilitating a timely and consistent evaluation.
Notified bodies may also, outside of the CECP process, seek supplementary scientific or clinical advice from the expert panels where additional clarification or input is deemed necessary to support the assessment of a BtX device.
Such proactive interaction and prioritisation should contribute to a more efficient conformity assessment, improved predictability for manufacturers, and ultimately to earlier access to safe and effective breakthrough technologies for patients and healthcare systems.
11.2 Support to NBs in the clinical assessment of BtMDs in line with MDR Article 106(11)
The notified body involved in the conformity assessment of a medical device which was granted BtMD status may seek advice from an expert panel in accordance with MDR Article 106(11). Before submitting such a request, the notified body should consult the manufacturer to inform them and in case further input into to the request is needed. Having regard to the limited capacity of the expert panels, notified bodies are advised to reach out to the EMA expert panel secretariat as early as possible to include an envisaged request for advice in the expert panels' planning.
The request for advice from the notified body will concern the clinical data required for the manufacturer's clinical evaluation, taking into account the considerations of this guideline. For that purpose, the notified body should put forward to the expert panels specific questions for which it seeks the panels' advice. Those questions should be based on a preliminary analysis of the clinical evaluation provided by the manufacturer.
The notified body should determine the timing of the consultation in agreement with the manufacturer depending on how it fits best in the overall conformity assessment procedure. The expectation is that the consultation will be conducted in parallel of the conformity assessment procedure with minimal impact on the overall timelines.
The expert panels will endeavour to provide their advice within 60 days, and to prioritise the devices that received a positive opinion regarding their BtX status.
11.3 Considerations for SMEs manufacturing BtX
In line with Annex VII, Section 1.2.8 of MDR and IVDR, notified bodies must ensure that their terms of service are fair and reasonable, taking into account the needs of SMEs. This is also of relevance to SMEs manufacturing breakthrough devices.
Therefore, in addition to the requirement to establish and publish transparent fee structures 20 that take into account the interest of micro, small, and medium enterprises 21 , notified bodies should consider adopting the following in the context of SMEs manufacturing BtX:
- Provide support and structured dialogue mechanisms to help navigate regulatory expectations,
- Establish procedures to facilitate the conformity assessment of breakthrough devices in a transparent and timely manner that is cost-effective and proportionate to the interests of SMEs.
11.4 Certification with Specific Conditions or Provisions
BtX for which the pre-market clinical evidence is deemed sufficient but needs to be completed or confirmed through PMCF/PMPF, are a good example where notified bodies can make use of the possibility to issue certificates with specific conditions or provisions. These conditions should be proportionate to the characteristics of the BtX, including its intended purpose, target population, duration of use, benefits and risks, clinical evidence, and any rare or latent risks or other aspects that may not have been fully characterised in the pre-market setting. Due consideration should be given to the guidance on clinical evidence and PMCF/PMPF in this document, as well as any relevant advice, opinions, or supports that have been provided to the BtX manufacturer, including scientific advice from EMA expert panels and regulatory advice from NCAs.
Specific conditions or provisions 20 may include, for example:
- Defined PMCF/PMPF activities, e.g., PMCF studies, within a specified period of time to generate additional clinical data,
- Enhanced surveillance and monitoring, including periodic milestone reporting on safety and performance,
- Informing users and patients (in the labelling, IFU, SSCP/SSP and/or other accompanying documentation) of:
- the BtX status of the device,
- the clinical evidence supporting the BtX,
- instructions to users on how to report incidents, complaints, and other clinical experience to the manufacturer.
- BtX manufacturer should inform the EMA/MDCG that they have achieved market access (see Section 14 for more details on Transparency).
11.5 Surveillance Activities by Notified Bodies
For breakthrough devices, notified bodies should re-assess the updated clinical evaluation reports and the results of any PMCF/PMPF studies, at an increased frequency that is proportionate to the level of clinical risk for the device.
The notified body should consider PMS data, including the main findings from PMCF/PMPF as part of the agreed surveillance activities and PSUR 22 evaluation and verify whether the device's benefit-risk profile continues to support the placing of the device on the market. As part of their surveillance activities and post-certification monitoring, notified bodies need to monitor compliance with any conditions/provisions that are binding for the manufacturer and associated with the certification decision, such as updates to clinical (performance) data at defined intervals. Where applicable, especially if listed as part of the conditions for certification, the notified body also needs to review the clinical evaluation that the manufacturer has updated based on its PMS, PMCF/PMPF.
When the conditions/provisions on the certificates are not fulfilled/met by the manufacturer, the notified body should consider the impact thereof on the certificate's validity, as specified in their procedures. Not fulfilling the conditions/provisions could ultimately lead to suspension or withdrawal of the certificate.
12. Roles of National Competent Authorities
Among their responsibilities, NCAs are responsible for assessing clinical investigations of MDs and clinical performance studies of IVDs, , market surveillance, enforcement, and
Medical Device Coordination Group Document coordination of regulatory activities at both national level and EU level. In the context of breakthrough devices, NCAs can play a role in:
- Supporting BtX manufacturers (especially SMEs) in understanding the requirements laid down by the MDR and IVDR and other potentially applicable legislation,
- Facilitating the priority assessment of clinical investigations and clinical performance studies of BtX requiring NCA authorisation, including coordinated assessments 23 where applicable,
- Communicate with MDCG, Expert Panels, and Notified Bodies in cases of emerging safety issues.
DAs are responsible overseeing notified body and can play a role within the scope of NCA oversight as designating authority, ensuring that surveillance activities agreed by notified bodies for BtMDs and BtIVDs reflect the risk profiles and data generation pathways of these devices and verification that SME interests are being appropriately reflected.
Several member states have established innovation offices or other forms of contacts to support manufacturers with their regulatory activities, including but not limited to information on compliance with the MDR and IVDR, and on the requirements surrounding clinical investigations and clinical performance studies Some NCAs may provide tailored regulatory advice and information to developers at national level. The EMA expert panel secretariat will act as the contact point for that request and liaise with the relevant authority/ies.
13. Funds to support innovation
Recognising that breakthrough devices often require significant early investment in regulatory strategy, clinical development and PMCF/PMPF, this section intends to provide a high-level overview of available instruments.
By leveraging both EU-wide programs and national initiatives, medical device companies can access a broad spectrum of funding opportunities to support their innovation and growth objectives.
13.1 National Funding Mechanisms
Many countries have designated agencies or contact points that provide guidance on available funding opportunities and application procedures.
13.2 European Funding mechanisms
At Union level, developers of breakthrough devices may be supported through:
The EU4Health Programme, particularly for technologies aligned with digital health, cancer, and rare diseases;
The European Innovation Council (EIC) Accelerator, which offers blended financing (grants + equity) to high-risk, high-impact innovations;
Horizon Europe health clusters and the Innovative Health Initiative (IHI) for collaborative R&D efforts;
The InvestEU Programme, which includes access to risk-sharing instruments for SME-led health technology innovation.
Enterprise Europe Network (EEN): EEN provides support for SMEs seeking to innovate and grow internationally, including advice on funding and access to finance.
14. Transparency of BtX
In the interest of transparency and the coordinated governance and oversight of the procedures described in this guidance, it is essential that all relevant stakeholders, including users, healthcare professionals and patients have appropriate access to information regarding the Btx status and evidence base.
The manufacturer should ensure that such transparency is achieved through the clear and consistent communication of relevant information in the device's labelling, instructions for use (IFU), Summary of Safety and Clinical Performance (SSCP) or Summary of Safety and Performance (SSP), and/or other accompanying documentation, as appropriate.
This information should include, as relevant:
- The designation of the device as a Breakthrough Technology (BtX)
- The EUDAMED registration details, ensuring public traceability of the device in accordance with the MDR/IVDR requirements;
- The SSCP or SSP, providing accessible and up-to-date information on the device's performance, safety, and clinical evidence, as made available through EUDAMED;
- The clinical investigation or performance study registration details (if applicable), including references to relevant public registries where information on ongoing or completed investigations is available;
- Instructions to users, health professionals, and patients on how to report incidents, complaints, and other clinical experiences directly to the manufacturer, as part of the device's post-market surveillance system.
The Commission will aim to develop a public dashboard to facilitate access to information on the number of BtX designated, those currently undergoing certification with the name and number of the notified bodies, those who have received certificates and have achieved market access.
Manufacturers and notified bodies are expected to proactively communicate relevant updates on the BtX status or supporting evidence through this publicly accessible dashboard, in line with applicable regulatory and confidentiality requirements.
Appendix A.1 - Table on BtX determination
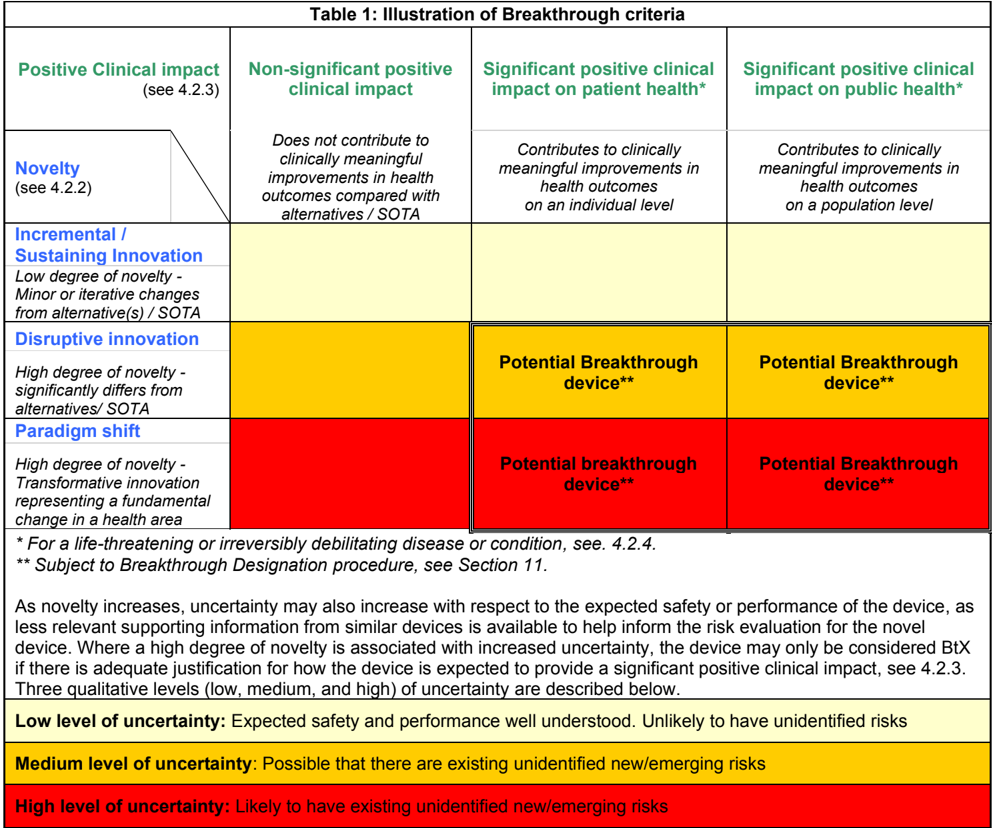
Appendix A.2 -Considerations on Clinical Investigations and Performance Studies for BtX
Study Design:
While randomized controlled trials (RCT) are often considered the preferred study design for clinical investigations of medical devices, this study design may not suit many BtX devices for example, where there are no alternatives or where available alternatives are not considered to offer similar patient benefit (lack of equipoise). In these cases, simpler methodological approaches may be acceptable if well justified. In cases where the state of the art may already provide satisfactory diagnostic or therapeutic options, a comparative study design may be more appropriate. The choice of design should be carefully considered and justified on a caseby-case basis in terms of its strengths and limitations (e.g. vulnerability to bias or confounders) and its ability to assess and confirm the positive clinical impact on patient and/or public health.
As an illustrative example, adaptive and sequential designs may be an appropriate design for confirmatory pre-market clinical investigation. These studies are based on interim analyses that are planned to be carried out over the course of the CI/PS.
An adaptive design may allow for potential changes in parameters (e.g. sample size requirements, randomization ratios, number of analyses) as the study progresses. However, care should be taken to ensure the integrity of the study is not compromised as a consequence of excessive interim analyses or adaptations. Where possible, all anticipated or potential adaptations of the study should be described in the CI/PS plan, based on anticipated results of the planned interim analyses.
A sequential design is based on interim analysis of the study's primary endpoints, and unlike adaptive studies, no adaptation of parameters is allowed. Instead, the stepwise methodology allows for continued, periodic analyses after each predefined group/cohort/stratum of patients reach their outcome, thus allowing for interim opportunities to determine whether there is sufficient evidence of clinical benefit or lack thereof.
Where appropriate, the findings from interim analyses may demonstrate sufficient clinical evidence for initial conformity assessment and market access. In this event, where possible and where there still is equipoise, the study should continue in the post-market setting to completion as planned in the CI/PS plan, to verify the finding from interim analysis, and to ensure appropriate follow-up for the study participants. Although adaptive and sequential methodologies can provide more flexibility, it is important to note that this approach can be vulnerable to type I (false positive) error. This potential for error should be considered when evaluating clinical data generated from these study designs.
The below table illustrates suggested study designs for certain high risk BtMD, as adapted from recommendations 24 developed by CORE-MD research consortium.
| Suggested study designs for Class III / implantable breakthroughMD* | |||||
|---|---|---|---|---|---|
| Pre-market CIs and studies | Pre-market CIs and studies | PMCFinvestigations& other activities** | PMCFinvestigations& other activities** | ||
| Initial/Early | Pre-market confirmatory | Post-market confirmatory | Long-term | ||
| Recommended | Case report(s) of first use | Comparative CI with concurrent matched controls, e.g., RCT | RCT vs SOTA / concurrent matched controls +/- blinding as appropriate | Registry-based study with enrolment of representative majority of patients | |
| Recommended | Comprehensive case series with prospective data collection | Single arm CI with all consecutive patients | Observational cohort study with concurrent matched controls | Registry-based study with enrolment of representative majority of patients | |
| Only if duly justified*** | Retrospective / post-hoc review of individual cases | Comparative CI with historical controls | Comparative CI with historical controls | Registry with unrepresentative recruitment |
- This table gives study design recommendations for certain high risk (Class III / implantable) interventional BtMDs. It is acknowledged that this table may not be applicable to certain BtMDs, such as lower risk (non-implantable, non-Class III) or non-therapeutic BtMDs. Similarly, this guidance is not foreseen as being directly applicable to BtIVDs.
** As described in Section 8 of this guidance, PMCF activities should be designed to confirm the safety and clinical performance of BtX throughout their expected lifetime. Thus, the duration of follow up of all patients enrolled in a PMCF study should be appropriate for the lifetime evaluation of the safety and clinical performance of the BtX.
*** The study designs listed in the final row should only be considered if it can be duly justified as to their appropriateness, with justification as to why the recommended study designs are not suitable for the BtMD in question.
Objectives
It may be suitable for the primary objectives of their pre-market clinical investigations of BtMD to focus on the assessment of short- to medium-term performance and patient safety
Pre-market performance studies of BtIVD should focus on establishing a base level of evidence to confirm/establish the safety and analytical performance. It is acknowledged that challenges may exist with respect to the generation of clinical performance data.
For PMCF investigations of medical devices, the objectives should aim to evaluate the overall safety, performance, and clinical benefit of the device throughout its expected lifetime, with a suitable focus on long-term endpoints.
For PMPF studies, the focus should be on confirming the safety and performance through the use of planned activities, for example, long term studies, EQA schemes, real world evidence collection etc. (See section 8)].
Endpoints:
Although disease-specific clinical endpoints remain the standard, such endpoints may not be sufficiently established, understood, or validated in the context of BtX. In this regard, appropriately validated surrogate endpoints can be considered, if justified. In these cases, relevant disease-specific clinical endpoints should subsequently be investigated, where possible, in the post-market setting, e.g., in PMCF investigations / PMPF studies.
When selecting endpoints, due consideration should be given with respect to how the endpoints can be used to confirm the claimed positive clinical impact of the BtX on patient and/or public health. When considering positive clinical impact on patient health, it is important to identify and consider the priorities and needs from the perspective of the patient. To that end, patient-reported outcomes (PROs) and other patient-centric measures can be considered for inclusion e.g. as secondary/exploratory outcomes and endpoints or as primary endpoints for evaluating clinical benefit in exceptional circumstances where it can be justified that other endpoints cannot be collected in the target population. When considering the inclusion of PROs and other clinically relevant endpoints, input from independent patients' representatives should be sought, if available and appropriate.
Choice of comparator/control
As previously discussed, the choice of comparator is dependent on several factors including equipoise and the availability (or absence thereof) of suitable alternatives.
In general, a concurrent active comparator is the preferred option if appropriate, where the active comparator is an appropriate alternative or represents the current state of the art. Sham or placebo may be appropriate controls, however subjects in these arms must receive the state of the art in management of their condition - i.e., they must receive care at least to the same level as the care they would receive in normal clinical practice.
In some circumstances, data from open-label studies with historical controls might be acceptable, however this design must be well justified, including justification as to why other study designs are not appropriate.
Clinical
/Performance
Evaluation
Certification
Appendix A.3 - Illustrative overview of support mechanisms across different stages of development
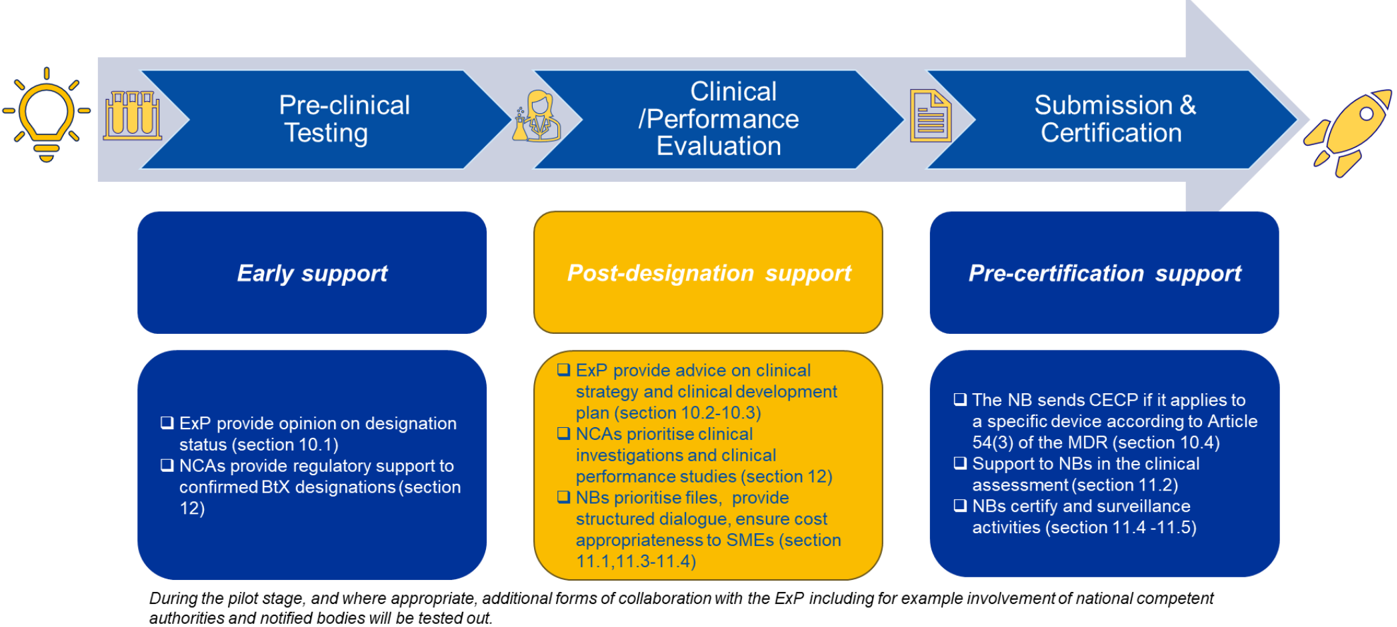
Submission &
Appendix A.4 - Opportunities and benefits which may result from BtX designation
Potential opportunities may include the following:
Scientific and Regulatory advice
Priority in EMA expert panel early scientific advice
Scientific advice to non-Art.61(2) BtMD and BtIVD (pilot)
Regulatory advice
Early and priority structured dialogue with NBs
CI/PS supports
Dedicated CI/PS pre-submission meetings
Supports/advice for early feasibility studies
Priority selection in CI/PS coordinated assessments (MDR A.78 / IVDR A.70)
Proportionate fees for CI/PS authorisations
Conformity assessment
Enhanced predictability for conformity assessment timelines and expectations
Priority structured dialogue with NB
Proportionate fees for BtX conformity assessment
Accelerated conformity assessment timelines
Special considerations re pre-clinical evaluation
Certification with conditions to facilitate safe, early market access
Pre-clinical and clinical evaluation
Additional considerations re standards, pre-clinical evidence for BtX
Guidance re meeting pre-clinical GSPRs in the absence of well-defined standards for BtX,
Guidance re clinical development strategies for BtX, including considerations for suitable CI designs in early, pre-market confirmatory, and post-market stages see Appendix A.2
Guidance on sufficient clinical evidence for conformity assessment
Increased role of PMCF for post-market confirmation of safety and performance
Cross-body recognition of BtX designation status for:
HTA assessments
EU and national grants and financial supports
Footnotes
- ' Everyone has the right of access to preventive health care and the right to benefit from medical treatment under the conditions established by national laws and practices. A high level of human health protection shall be ensured in the definition and implementation of all Union policies and activities. '
- See also Commission guidance for expert panels on CECP (2020/C 259/02)
- For IVDs the application of the device in laboratory practice may also be considered.
- E.g., certain class D IVDs, per IVDR Article 48(6)
- Per Commission guidance for expert panels on CECP (2020/C 259/02)
- As defined in Article 2(66) MDR and Article 2(69) IVDR.
- Certain IVDs may be considered to provide benefit for life-threatening conditions either by being explicitly stated in the IVDR classification rules (Annex VIII), as outlined in MDCG 2020-16, or by being determined based on the nature of the condition itself.
- For BtMD, clinical evidence must be provided by clinical data, whereas for BtIVD, clinical evidence must be provided by scientific validity, analytical performance, and clinical performance data, per MDR Article 61(1) and IVDR Article 56(1), respectively.
- In accordance with MDR Chapter VI and Annex XIV and IVDR Chapter VI and Annex XIII.
- Clinical data for MDs; scientific validity, analytic performance, and clinical performance data for IVDs.
- Including non-clinical/pre-clinical data, clinical data for MDs, and scientific validity, analytical performance, and clinical performance data for IVDs.
- As defined in MDR Article 2(53) for MDs, and IVDR Article 2(37) for IVDs.
- MDCG 2025-6 - FAQ on Interplay between the Medical Devices Regulation (MDR) & In vitro Diagnostic Medical Devices Regulation (IVDR) and the Artificial Intelligence Act (AIA).
- Per IVDR Article 56(4).
- See guidance on 'Post-market surveillance of medical devices and in vitro diagnostic medical devices'.
- Per MDR Article 56(3) / IVDR Article 51(3).
- See IMDRF Principles of International System of Registries Linked to Other Data Sources and Tools (2016).
- Per Chapter VI and related Annexes of the MDR and IVDR, as applicable.
- Per section 10.1.
- In accordance with MDR Article 50 and IVDR Article 48.
- As outlined in MDCG 2023-2.
- Pursuant to MDR Article 86 / IVDR Article 81.
- Per MDR Article 78 and IVDR Article 74.
- See Recommended methodologies for clinical investigations of high-risk medical devicesConclusions from the European Union CORE-MD Project (Lancet Reg Health Eur 2025).