MDCG 2021-24 rev.1:医疗器械分类指南
修订说明
本文为 Rev.1 版(2026年4月),替代2021年10月原版。主要变更包括:"临床操作"术语澄清、规则8新增实务说明、规则10和12分类表更新。
文件编号:MDCG 2021-24 rev.1
发布日期:2026年4月
适用法规:EU MDR 2017/745
官方PDF:下载
公告链接:EC Public Health
Rev.1 主要变更
- 第3.1.4节(植入器械):"手术操作"(surgical procedure)替换为"临床操作"(clinical procedure)——明确通过非手术技术(套管、导管、针头)引入的器械在满足30天标准的情况下也可定义为植入器械。
- 规则8:新增2条实务说明(总计7条):
- 说明1:第52(4)条豁免仅涉及合规评估路径,不影响分类结论。
- 说明7:脊柱接触范围扩展至颈椎到尾椎的所有骨结构(包括棘突和横突)。
- 规则10和12:分类表更新新增示例(手术灯、眼底镜、检眼镜、弹性泵、球囊泵输注)。
- 规则9:结构重组以提高清晰度。
- 规则16:灭菌器分类指南更新。
- 规则22:除颤器附件澄清。
文件目的
本指南提供了EU MDR附件VIII分类规则的实践解读,帮助制造商和主管机构正确确定医疗器械的风险分类(Class I、IIa、IIb、III)。
EU MDR 分类体系概述
分类规则框架
EU MDR附件VIII包含22条分类规则,分为以下几类:
| 规则类别 | 规则编号 | 适用器械类型 |
|---|---|---|
| 非有源器械 | 规则1-4 | 非有源、非植入式器械 |
| 有源器械 | 规则9-13 | 有源器械(含软件) |
| 特殊规则 | 规则14-22 | 特定类别器械 |
| 植入式器械 | 规则5-8 | 植入式和长期接触器械 |
分类等级
| 分类 | 风险等级 | 典型示例 |
|---|---|---|
| Class I | 低风险 | 绷带、手术器械(非无菌/非测量) |
| Class IIa | 中低风险 | 助听器、隐形眼镜 |
| Class IIb | 中高风险 | 呼吸机、输液泵 |
| Class III | 高风险 | 心脏瓣膜、冠状动脉支架、植入式神经刺激器 |
关键分类规则解读
规则1 — 非有源非植入式器械(基础规则)
Class I(默认):所有非有源、非植入式器械,除非适用其他规则。
Class IIa:
- 短期使用(> 60分钟,≤ 30天)的有创器械
- 与受伤皮肤接触的器械(如伤口敷料)
规则9 — 有源治疗器械
- Class IIa:有源治疗器械(低风险能量传递)
- Class IIb:有源治疗器械(高风险能量传递,如放射治疗设备)
- Class III:有源植入式器械
规则11 — 软件
软件分类须考虑:
- 软件的预期目的
- 错误决策对患者的潜在影响
Class I:不影响临床决策的软件(如行政管理软件)
Class IIa:影响临床决策但错误影响可逆的软件
Class IIb:影响临床决策且错误影响可能严重但可逆
Class III:影响临床决策且错误可能导致不可逆严重后果
规则22 — 附件XVI产品
附件XVI列出的非医疗目的产品(如接触镜、美容植入物)须按照相应分类规则分类。
分类决策流程
确定器械预期目的
↓
是否为有源器械?
↓ ↓
是 否
↓ ↓
应用规则9-13 是否为植入式器械?
↓ ↓
是 否
↓ ↓
应用规则5-8 应用规则1-4
↓
检查特殊规则14-22
↓
确定最终分类(取最高分类)分类争议处理
若制造商对分类有疑问:
- 向所在成员国主管机构咨询
- 主管机构可要求MDCG提供意见
- 欧盟委员会可发布实施措施明确分类
相关文件
官方文件全文
MDCG 2021-24 Guidance on classification of medical devices
October 2021
This document has been endorsed by the Medical Device Coordination Group (MDCG) established by Article 103 of Regulation (EU) 2017/745. The MDCG is composed of representatives of all Member States and it is chaired by a representative of the European Commission.
The document is not a European Commission document and it cannot be regarded as reflecting the official position of the European Commission. Any views expressed in this document are not legally binding and only the Court of Justice of the European Union can give binding interpretations of Union law.
MDCG 2021- 24
1 Purpose of medical device classification
The classification of medical devices in use by the EU medical device legislation is a risk-based system taking into account the vulnerability of the human body and the potential risks associated with the devices. This approach uses a set of criteria that can be combined in various ways in order to determine classification, e.g. duration of contact with the body, degree of invasiveness, local vs. systemic effect, potential toxicity, the part of the body affected by the use of the device and if the device depends on a source of energy. The criteria can then be applied to a vast range of different medical devices and technologies. These are referred to as the 'classification rules' and are set out in Annex VIII of Regulation (EU) 2017/745 on medical devices (MDR). They correspond, to a large extent, to the classification rules established by the International Medical Device Regulators Forum (IMDRF) in the guidance document GHTF/SG1/N77:2012 1 .
2 Practical relevance of classification
The purpose of this chapter is to provide a general overview on the impact of the classification of medical devices on different aspects of the device compliance with the legal requirements. The explanations provide some simplified concepts and are not exhaustive. For details see the MDR and related additional guidance 2 .
2.1 General requirements
Irrespective of the class of the device, all devices must comply with all relevant obligations of the MDR. However, some requirements depend on the device classification.
For example, the devices must:
- meet the general safety and performance requirements, including the requirements regarding the information to be supplied by the manufacturer (Annex I of the MDR);
- be subject to the reporting requirements under the medical device vigilance system;
- be CE marked (except custom-made devices and devices intended for clinical investigation, in which case they should comply with the provisions of respectively Art. 52.8 and Annex XIII or Articles 62 - 80, 82 and Annex XV);
- be assigned a Unique Device Identifier (UDI) number and be registered in the electronic system, in accordance with MDR Article 29;
- if the device is implantable, be supplied with an implant card and information to the patient in accordance with Article 18.
MDCG 2021- 24
According to MDR Article 51 devices are divided into the following classes I, IIa, IIb and III, taking into account the intended purpose of the devices and their inherent risks. Classification is to be carried out in accordance with Annex VIII to the MDR. In addition, and according to Article 52(7)(a), (b) and (c), Class I devices can be further subdivided into Is - sterile condition, Im - measuring function and Ir - reusable surgical.
The technical documentation 3 to be drawn up by the manufacturer must include the risk class of the device and the justification for the classification rule(s) applied in accordance with Annex VIII of the MDR.
Sections 2.2-2.6 give an overview of some requirements that depend on the class of the device. For detailed and exhaustive provisions on each topic, refer to the MDR, as well as to dedicated guidance where available 4 .
Annex XVI products should be classified in accordance with the classification rules in Annex VIII of the MDR and taking into account possible provisions within the relevant implementing acts covering Annex XVI devices.
2.2 Conformity assessment
Conformity assessment is the process demonstrating whether the requirements of the MDR relating to a device have been fulfilled. Demonstrating conformity is in the first instance the responsibility of the manufacturer and for most devices classes the conformity is then assessed by a notified body. The higher the class of the device, the greater the involvement of a notified body in conformity assessment. Annex I (general safety and performance requirements) and Annexes II (technical documentation) and III (technical documentation on post-market surveillance) apply to all devices regardless of class. Further relevant conformity assessment procedures (laid down in Annexes IX to XI) will depend on the class of the device. For some classes, the manufacturer has a choice of more than one procedure. Conformity assessment is described in MDR Article 52.
Custom-made or investigational devices falling into any class have their own provisions: Annex XIII for custom-made devices and Article 82 for investigational devices. For class III implantable custom-made devices, the manufacturer also needs to apply either Chapter I of Annex IX or Part A of Annex XI.
2.3 Clinical evaluation and investigation
For any device regardless of class, the manufacturer must ensure the general safety and performance requirements are satisfied (MDR Article 5, MDR Annex I). This includes carrying out a clinical evaluation (MDR Article 5 (3), MDR Article 61, MDR Annex XIV. For implantable devices and class III devices, a premarket clinical investigation is compulsory, with some exceptions such as modifications of an existing device, demonstrated equivalence to CE-marked device, placed on the market under Directive 90/385/EEC or Directive 93/42/EEC for which sufficient clinical data is already available, and specific exemptions laid down in Article 61(6)(b). The conditions for starting a clinical investigation vary
MDCG 2021- 24 depending on the class of the device (see MDR Article 70(7) and Article 78). According to Article 61(10), if demonstration of conformity with Annex I requirements based on clinical data is not deemed appropriate, the manufacturer shall justify this in the technical documentation.
For class III implantable devices and class IIb active devices intended to administer or remove a medicinal product, the notified body must also follow the clinical evaluation consultation procedure where certain documentation including the clinical evaluation report is submitted for review by expert panels (MDR Article 54 and Section 5.1 of Annex IX). It must notify the Member State competent authorities of the certificates it has granted for these types of devices (MDR Article 55). The manufacturer may consult an expert panel on their clinical development strategy prior to performing the clinical evaluation and/or investigation (MDR Article 61(2)). See also MDCG Guideline 2019-3 5 for interpretation of Article 54.
For implantable devices and class III devices, other than custom-made or investigational devices, the manufacturer must update the postmarket clinical follow-up evaluation report as it will serve an input for the writing of the Periodic Safety Update Report, and, if indicated, the summary of safety and clinical performance 6 (MDR Article 32).
2.4 Post-market surveillance
The manufacturer must update the clinical evaluation with clinically relevant information coming from post-market surveillance, in particular the post-market clinical follow-up.
For class I devices, including sterile, those with a measuring function and the reusable surgical instruments, the manufacturer must prepare and maintain a post-market surveillance report (MDR Article 85), which must be made available to the competent authorities on request.
For class IIa, IIb and III devices, the manufacturer must prepare a periodic safety update report for each device, and, where relevant, for each category or group of devices (MDR Article 86). This report must be updated at least annually for class IIb and III devices and at least every two years for class IIa devices .
2.5 Traceability
For class III implantable devices, economic operators and health institutions are obliged to have a record of the UDI of the devices they have supplied or with which they have been supplied (MDR Article 27).
For class II and III devices, the economic operator is obliged to provide information on the Member State(s) where the device is, or is to be, made available when registering the device (Annex VI Part A 2.4). In the case of implantable and class III devices, the economic operator must provide the summary of safety and clinical performance (Annex VI Part A 2.14). For single-use class I and IIa devices packaged and labelled individually, the UDI carrier does not have to appear on the packaging but must appear on a higher level of packaging (Annex VI Part C 4.3).
However, if the healthcare provider does not have access to the higher level of packaging, the UDI carrier must appear on the individual device packaging.
2.6 Instructions for use
Generally, instructions for use must be supplied together with the device. By way of exception, class I and IIa devices may be supplied without instructions for use if such devices can safely be used without the instructions and no other provisions of Annex I Section 23 state otherwise.
3 How to carry out classification
This section is aimed at presenting definitions and terms useful for the application of the classification principles and rules.
3.1 Basic terms and definitions
Relevant definitions in respect to the classification of devices are set out in Article 2 and Chapter I of Annex VIII of the MDR but the Regulation also contains explanation of further terms. These relevant terms and definitions for this guidance are collected below.
Specific medical purpose
The specific medical purpose is specified by the manufacturer from those listed in the indents of Article 2(1) MDR.
Duration of use
Transient
Normally intended for continuous use for less than 60 minutes.
Short term
Normally intended for continuous use for between 60 minutes and 30 days .
Long term
Normally intended for continuous use for more than 30 days.
In certain instances the duration of use for a product needs to be considered as the duration of effect. For instance, application of a topical cream to the skin may only take seconds to apply but the cream may remain in situ for many hours. The duration of use should therefore not be considered as the time taken to apply the product but rather the duration for which the product remains in or on the body.
Continuous use
In calculating the duration referred to in Section 1 of Chapter I of Annex VIII of MDR, continuous use means:
'(a) The entire duration of use of the same device without regard to temporary interruption of use during a procedure or temporary removal for purposes such as cleaning or disinfection of the device. Whether the interruption of use or the removal is temporary shall be established in relation to the duration of the use prior to and after the period when the use is interrupted or the device removed; and
(b) the accumulated use of a device that is intended by the manufacturer to be replaced immediately with another of the same type.'
For example, a scalpel may be used on the same patient throughout an operation that may last for several hours. The uninterrupted use for an intended purpose, i.e. cutting tissue, will normally not last for more than a few seconds at a time. Therefore a scalpel is a transient use device. However, where usage of a device is discontinued in order for the device to be replaced immediately by the same or an identical device ( e.g . replacement of a ureteric catheter) this shall be considered an extension of the continuous use of the device.
As another example, the overnight period when contact lenses are cleaned and disinfected is considered as a discontinuation of the device use. For the determination of the duration of use, only the specified time period of uninterrupted wear of the lens (e.g. 16 hours) needs to be taken into account 7 .
If it cannot be demonstrated that components of the device are totally discontinued in the interval between uses, this is also considered as an immediate replacement and an extension of the continuous use of the device.
Invasiveness
Invasive device
Any device which, in whole or in part, penetrates inside the body, either through a body orifice or through the surface of the body. A device that administers energy to the body should not be considered as invasive if only energy it emits penetrates the body and not the device itself.
Body orifice
Any natural opening in the body, as well as the external surface of the eyeball, or any permanent artificial opening, such as a stoma.
Injured skin or mucous membrane 8
An area of skin or a mucous membrane presenting a pathological change or change following disease, a wound or a scar.
Surgically invasive device 9
An invasive device which penetrates inside the body through the surface of the body, including through mucous membranes of body orifices with the aid or in the context of a surgical operation; and a device which produces penetration other than through a body orifice.
The term surgical operation used in this definition includes all clinical interventional procedures in which a device is placed into the body through the surface of the body. A surgically invasive device always implies that it enters through an artificially created opening. This can be a large opening, such as a surgical incision, or it can be a pinprick opening made by a needle. Therefore surgical gloves and needles used with syringes are surgically invasive.
In this context the following should be noted:
- a surgically created stoma used in urostomy, colostomy and ileostomy or permanent tracheostomy is considered to be a body orifice; therefore devices introduced into such a stoma are not surgically invasive.
- in contrast, a surgically created opening to allow access to the circulatory system should not be considered to be a body orifice. Devices introduced into such an opening are surgically invasive.
The concept of surgically invasive should be understood to cover also liquids that are in invasive contact with organs, tissues or other parts of the body if the access for such liquids is through a surgically created opening.
For a device that administers a substance, such a substance must be assessed in its own right (e.g. substances administered by a jet injector).
Reusable surgical instrument 10
An instrument intended for surgical use in cutting, drilling, sawing, scratching, scraping, clamping, retracting, clipping or similar procedures, without a connection to an active device and which is intended by the manufacturer to be reused after appropriate procedures such as cleaning, disinfection and sterilisation have been carried out.
Implantable device 11
Any device, including those that are partially or wholly absorbed, which is intended:
- to be totally introduced into the human body, or
- to replace an epithelial surface or the surface of the eye, by clinical intervention and which is intended to remain in place after the procedure.
MDCG 2021- 24
Any device intended to be partially introduced into the human body by clinical intervention and intended to remain in place after the procedure for at least 30 days shall also be deemed to be an implantable device.
A 'procedure' must be understood in this context to include the surgical procedure during which the implant is placed into the body and the immediate post-operative care that is associated with the procedure. The 'procedure' does not extend to the conclusion of the therapeutic treatment, e.g. the removal of an implant must be considered to be another 'procedure'. Thus a plate used to fix a fractured bone and which remains in place after the procedure for at least 30 days is an implant even if it is taken out after the fracture has healed. In this case the placing of the plate and its explantation are two different surgical procedures.
Sometimes partially implanted devices are deemed to be implants. For instance, if an operation is carried out specifically to place an infusion port into the body which would remain in place for at least 30 days after the procedure, such an infusion port would be an implant. However, a non-tunnelled central venous catheter which is intended for use for temporary vascular access and intended to be removed after 7 - 10 days is not an implantable device. Nor would a suture used for skin wound closure that is intended to be taken out prior to 30 days be considered an implant.
Critical anatomical locations
For the purposes of the MDR, ' Central circulatory system' 12 means the following blood vessels: arteriae pulmonales, aorta ascendens, arcus aortae, aorta descendens to the bifurcatio aortae, arteriae coronariae, arteria carotis communis, arteria carotis externa, arteria carotis interna, arteriae cerebrales, truncus brachiocephalicus, venae cordis, venae pulmonales, vena cava superior and vena cava inferior.
For the purposes of the MDR, ' Central nervous system ' 13 means the brain, meninges and spinal cord.
Active medical devices
Active device means any device, the operation of which depends on a source of energy other than that generated by the human body for that purpose, or by gravity, and which acts by changing the density of or converting that energy. Devices intended to transmit energy, substances or other elements between an active device and the patient, without any significant change, shall not be deemed to be active devices 14 .
The concept act by converting energy includes conversion of energy in the device and/or conversion at the interface between the device and the tissues or in the tissues. Electrodes intended for E.C.G. or E.E.G are normally not considered active devices because they do not normally act by conversion of energy.
MDCG 2021- 24
The application of energy from the human body for the purpose of operating a device does not make the device 'active' unless that energy is stored within the device for subsequent release. For instance, energy generated by human muscle and applied to the plunger of a syringe (thus causing a substance to be delivered to a patient) does not make this syringe an active device. However, if a drug delivery system depends upon manual winding to preload a spring which is subsequently released to deliver a substance, then the device incorporating the spring is an active device. Another example of an active device are elastomeric pumps, where the energy from the human body is stored in the stretched elastomer layer.
Medical devices using prestored gases and/or vacuum as a power source are regarded as active devices, as long as they fulfil both the criteria under the definition e.g. gas mixers with anaesthesia machines, aerosol pain relief sprays with a pre-stored propellant gas supply and gas-powered suction pumps.
Heating/cooling pads intended only to release stored thermal energy are not active devices because they do not act by conversion of energy. However, heating/cooling pads which act by chemical action (e.g. exothermic or endothermic reaction) are active devices as they are converting chemical energy into heat and/or vice versa .
The concept of significant change for energy includes changes in the nature, level and density of energy (see Rule 9). This means that for instance an electrode is not considered an active device under this classification system as long as the energy input is intended to be the same as the energy output. Resistance in a wire that causes minor changes between input and output cannot be considered to constitute 'significant change'. However, electrodes used in electrosurgery for cutting tissues or cauterisation are active devices because their operation depends on energy provided by a generator and their action is achieved by conversion of energy at the interface between the device and the tissue or in the tissue.
Software is also an active device 13 . Software should be reviewed not only in the context of Rule 11 15 .
Active therapeutic device means any active device used, whether alone or in combination with other devices, to support, modify, replace or restore biological functions or structures with a view to treatment or alleviation of an illness, injury or disability. 16
Active device intended for diagnosis and monitoring means any active device used, whether alone or in combination with other devices, to supply information for detecting, diagnosing, monitoring or treating physiological conditions, states of health, illnesses or congenital deformities 17 . A device is considered to allow direct diagnosis when it provides the diagnosis of the disease or condition in question itself or when it provides decisive information for the diagnosis 18 .
Devices with a measuring function
The following criteria, if fulfilled together, indicate that a device has a measuring function:
a) The device is intended by the manufacturer to measure:
quantitatively a physiological or anatomical parameter, or
a quantity or a quantifiable characteristic of energy or of substances (including medicinal products) delivered to or removed from the human body. Spoons or plastic syringes co-packed with medicinal products and used to measure a quantity of that medicinal product to be administered to the patient are in this category. Devices for the delivery of liquid to the human body without graduation or scale (e.g. medicine spoons, cups, droppers without graduation or scale or display of measuring unit) are not in this category 19 .
b) The result of the measurement:
is displayed in legal units or other acceptable units within the meaning of Directive 80/181/ECC 20 , or
is compared to at least one point of reference indicated in legal units or other acceptable units in compliance with the mentioned directive..
c) The intended purpose implies accuracy, claimed explicitly or implicitly, where a non-compliance with the implied accuracy could result in a significant adverse effect on the patient's health and safety.
The expression ' claimed implicitly ' covers cases where the user, on the basis of the designation of the device or of its accompanying documents, or on the basis of the common use, is entitled to expect accuracy where the accuracy of the measurement has an impact on the diagnosis or therapy of the patient.
Measuring activities during the manufacturing process including those for calibration purposes are not covered and do not imply a measuring function of the manufactured device.
Systems and procedure packs
System and procedure packs is described in MDR Article 22. They can combine medical devices, in vitro diagnostic medical devices, and other products which are in conformity with legislation that applies to those products, only where they are used within a medical procedure or their presence in the system or procedure pack is otherwise justified. In this case of demonstrated legal conformity of each component, the systems or procedure packs no not need to bear themselves an additional CE marking but they must bear the name, registered trade name or registered trade mark of the person who combines the products as well as the address at which that person can be contacted.
MDCG 2021- 24
A procedure pack means a combination of products packaged together and placed on the market with the purpose of being used for a specific medical purpose 21 . A system means a combination of products, either packaged together or not, which are intended to be interconnected or combined to achieve a specific medical purpose 22 .
Where the system or procedure pack incorporates devices which do not bear the CE marking or where the chosen combination of devices is not compatible in view of their original intended purpose, or where the sterilisation has not been carried out in accordance with the manufacturer's instructions the system or procedure pack shall be treated as a device in its own right and shall be subject to the relevant conformity assessment procedure pursuant to MDR Article 52. The classification is determined by the intended use of the system or procedure pack. The natural or legal person that combines the devices must assume the obligations incumbent on manufacturers.
For such combinations, including different devices, the classification is normally determined by the intended use. In those cases where the intended use of the final device is not specific enough to determine the classification, the classification of the device is at the level of the highest classified device included, taking into account the new intended use of the device.
Other terms
This section gives notes on other terms used in Annex VIII of the MDR:
Systemic absorption : The process by which substances or their metabolites enter the body (e.g. by crossing mucous membranes) and are distributed into the body via the blood and/or lymphatic system.
Wholly or mainly absorbed: The term 'absorption' in the context of implantable devices refers to the degradation of a material within the body and the metabolic elimination of the resulting degradation products from the body. It does not apply to those substances that are excreted without modification from the body, e.g. insufflation gases for the abdominal cavity or laparoscopic and endoscopic procedures.
Local dispersion : The condition by which substances remain in a specific site without being distributed into the body via the blood and/or lymphatic system.
Medicine / medicinal product : According to the definition given in Directive 2001/83/EC:
'(a) Any substance or combination of substances presented as having properties for treating or preventing disease in human beings; or
(b) Any substance or combination of substances which may be used in or administered to human beings either with a view to restoring, correcting or modifying physiological functions by exerting a pharmacological, immunological or metabolic action, or to making a medical diagnosis.'
MDCG 2021- 24
A ' nanomaterial ' means a natural, incidental or manufactured material containing particles in an unbound state or as an aggregate or as an agglomerate and where, for 50% or more of the particles in the number size distribution, one or more external dimensions is in the size range 1-100 nm; Fullerenes, graphene flakes and single-wall carbon nanotubes with one or more external dimensions below 1 nm shall also be deemed to be nanomaterials according to MDR, Article 2(18). Related definitions on 'particle', 'agglomerate' and 'aggregate' are also included in the MDR Article 2(19-21). The definitions on nanomaterial and the related terms were taken from Commission Recommendation 2011/696/EU on the definition of nanomaterials 23 . Guidance on terms and concepts used in the definition can be found in a report from the European Commission's Joint Research Centre. 24
Derivative means a non-cellular substance extracted from human or animal tissue or cells through a manufacturing process. The final substance used for manufacturing of the device in this case does not contain any cells or tissues.
3.2 Application of the classification rules
Before applying the classification rules, the manufacturer should first determine if the product concerned, based on its specific medical purpose , falls in the scope of the MDR as medical device, accessory for a medical device (Article 2 MDR), medical device part or component for replacement (Article 23(2) MDR) or as a device without an intended medical purpose listed in Annex XVI.
It is the intended and not the accidental use of the device that determines the class of the device. For instance, a suture organiser that is intended to keep suture threads used in open heart surgery in the correct order should not be considered as an invasive device if it is intended to be kept outside the patient. Similarly, if a healthcare professional or others uses the device in a manner not intended by the manufacturer, this does not change the class of the device for the purpose of conformity assessment. However, if the normal clinical use of the device changes in time with evolving clinical practice such that the intended purpose and classification of the device changes, this should be addressed by the manufacturer and the conformity of the device assessed for the new intended purpose. It is the intended purpose assigned by the manufacturer to the device that determines the class of the device and not the class assigned to other similar products. For instance, two sutures that have the same composition may well have different intended purposes.
In case several rules, or if, within the same classification rule, several sub-rules, apply to the same device based on the device intended purpose, the strictest rule and sub-rule resulting in higher classification will apply.
In terms of further clarification of the classification rules, the elements listed below should also be considered.
How to use the rules
The manufacturer must take into consideration all the rules in order to establish the proper classification for its device. The strictest rule and sub-rule resulting in the highest classification determines the class 25 . It is quite conceivable for instance that one of the general rules that is not specific to active devices nevertheless applies to such a device. The intended purpose and all the device characteristics must be taken into consideration. The characteristic or combination of characteristics in accordance with the intended purpose of the device that falls into the highest class determines the class for the device as a whole.
In addition to the classification rules set out in Annex VIII of MDR, the manufacturers must also take account of any applicable legal acts and consider guidance documents which may support the classification of their device.
Practical example
A simple wound drainage system has usually three components that must be taken into consideration: the cannula, the tubing and the collector unit. If the system is sold without a cannula, then the classification of the cannula does not need to be taken into account. It is assumed here that the system is used for short term duration, i.e. that uninterrupted intended use is more than 60 minutes and less than 30 days. It is furthermore assumed that the collected liquids are not intended to be reinfused into the body nor reprocessed for eventual reinfusion and that the device is not intended to be connected to a powered suction system.
| Intended uses | Rule | Class |
|---|---|---|
| Surgically invasive cannula to reach a wound site in the pleural cavity to drain the cavity | 7 | IIa |
| Non-invasive tubing to evacuate body liquids towards the collector. | 1 | I |
| Non-invasive collector to receive the body liquids. | 1 | I |
The clear conclusion here is that the manufacturer would have a choice of applying class IIa to the whole device or carrying out separate conformity assessment procedures for the cannula on one hand and the tubing and collector on the other hand.
3.3 Handling of interpretational problems
It is recognised that although the existing rules will adequately classify the vast majority of existing devices, a small number of devices may be more difficult to classify.
As soon a notified body needs to be involved, any dispute between the manufacturer and the notified body concerned, arising from the application of Annex VIII, may be referred for a decision to the competent authority of the Member State in which the manufacturer (or its authorized representative) has its registered place of business. In cases where the manufacturer has no registered place of business in the Union and has not yet designated an authorised representative, the matter shall be referred to the competent authority of the Member State in which the authorised representative referred to in the last indent of point (b) of the second paragraph of Section 2.2 of Annex IX has its registered place of business. Where the notified body concerned is established in a Member State other than that of the manufacturer, the competent authority must adopt its decision after consultation with the competent authority of the Member State that designated the notified body. The competent authority of the Member State in which the manufacturer has its registered place of business will notify the MDCG and the Commission of its decision. The decision can be made available upon request 26 .
Outside this regulatory procedure, competent authorities may refer on an ad hoc and voluntary basis complex classification cases for discussion at the Borderline and Classification Working Group of the MDCG. Agreement positions on classification reached by this Working Group are published for reference in the Manual on Borderline and Classification.
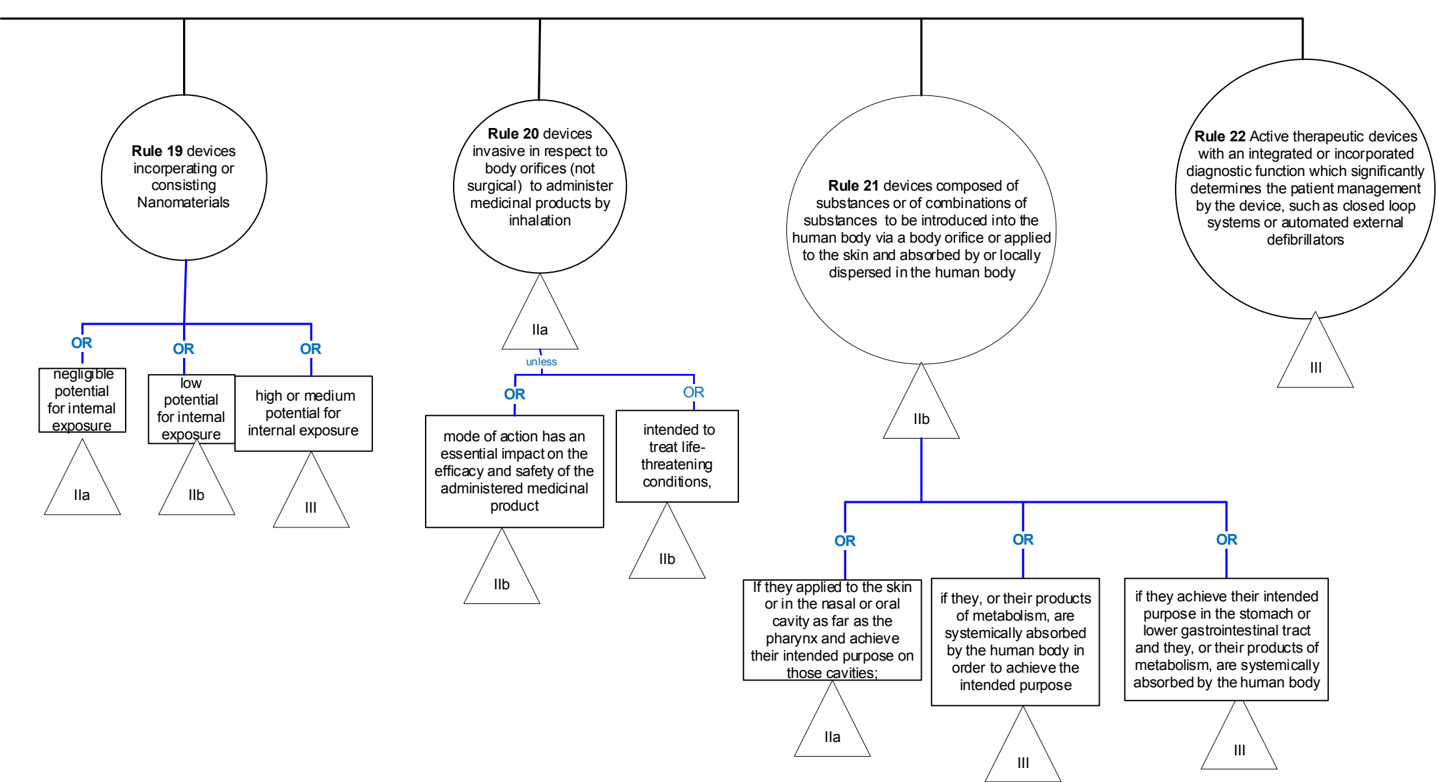
4 Explanations of individual rules
4.1 Graphical summary
Non-invasive devices
NON-INVASIVE DEVICES
ATTENTION: to special rules (14 to 22)
Invasive devices
Invasive devices
Invasive devices
Invasive devices
Invasive devices
Active devices
ACTIVE DEVICES
ACTIVE DEVICES
Special rules
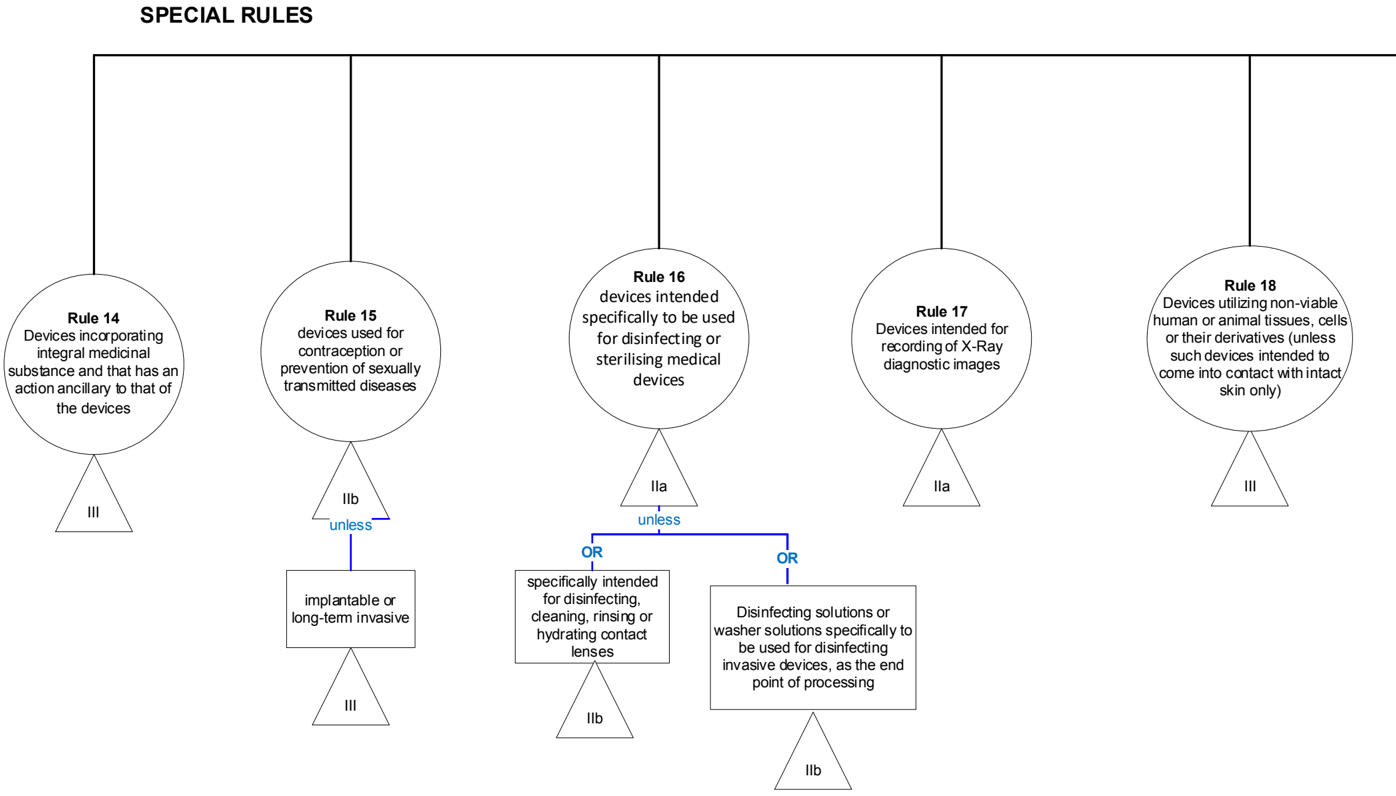
SPECIAL RULES
MDCG 2021- 24
4.2 General explanation of rules/practical issues/examples
The examples provided do not imply that the products are a priori qualified as devices. Classification rules apply after the qualification of the device has been established.
Non-invasive devices
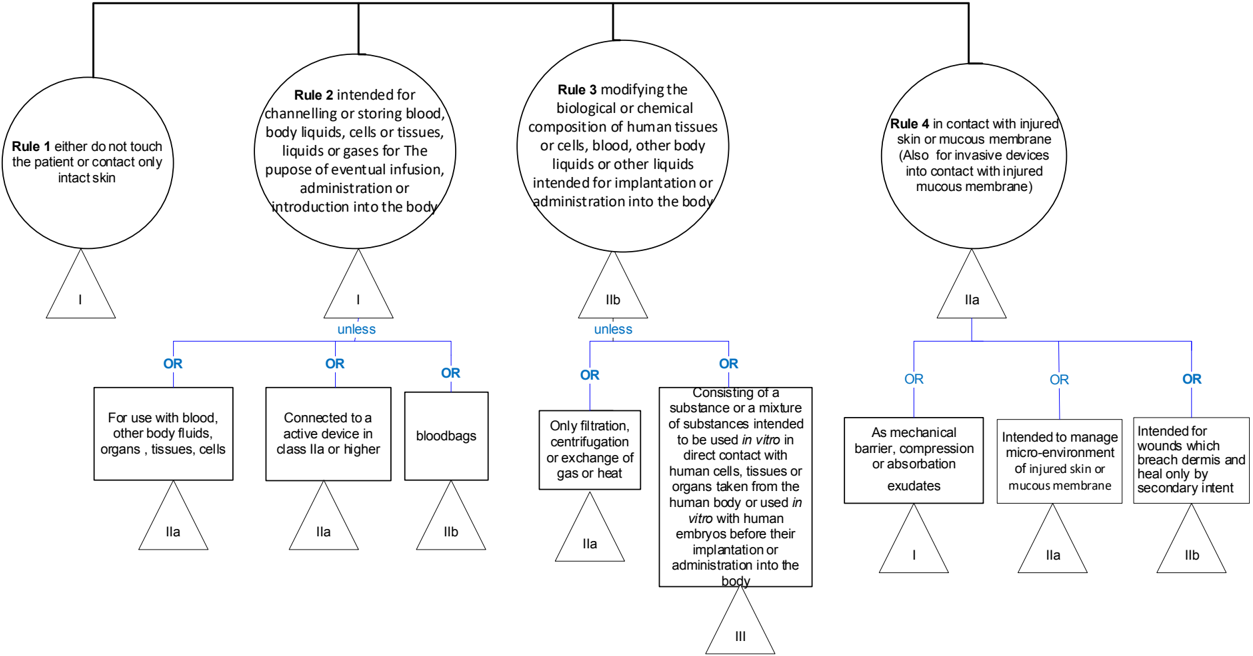
Rule 1 - Devices that either do not come in direct contact with the patient or contact intact skin 1 only
General explanation of the rule
This is a fall-back rule applying to all devices that are not covered by a more specific rule.
This is a rule that applies in general to devices that come into contact only with intact skin or that do not come in direct contact with the patient.
| Class | Rule 1 | Examples |
|---|---|---|
| I | All non-invasive devices are classified as class I, unless one of the rules set out hereinafter applies | Devices intended in general for external patient support ( e.g. hospital beds, patient hoists, walking aids, wheelchairs, stretchers , dental patient chairs) Body liquid collection devices intended to be used in such a way that a return flow is unlikely (e.g. to collect body wastes such as urine collection bottles, incontinence pads or collectors used with wound drainage devices). They may be connected to the patient by means of catheters and tubing Devices used to immobilise body parts and/or to apply force or compression on them ( e.g. non-sterile dressings used to aid the healing of a sprain, plaster of Paris, cervical collars, gravity traction devices, compression hosiery) Corrective spectacle frames (i.e. glasses) and lenses in frames Stethoscopes Eye occlusion plasters Incision drapes Non-invasive conductive gels i.e. ultrasound gels 2 Non-invasive electrodes (electrodes for EEG or ECG) Permanent magnets for removal of ocular debris Wheelchairs pushed by hand |
Practical issues of classification
Some non-invasive devices are indirectly in contact with the body and can influence internal physiological processes by storing, channelling or treating blood, other body liquids or liquids which are returned or infused into the body or by generating energy that is delivered to the body. These are handled by other rules because of the hazards inherent in such indirect influence on the body.
Note 1: Intact skin includes the skin around an established stoma unless the skin is breached. Signs of breached skin include, but not limited to, tears, erythema, oedema, weeping and infection. The definition of intact skin must apply for the continuous use of the device.
Note 2: Ultrasound gels are not to be absorbed or locally dispersed within the body at the site of action in order to achieve their intended purpose.
Rule 2 - Channelling or storing for eventual administration
General explanation of the rule
This rule applies to non-invasive devices intended for channeling or storing blood, body liquids, cells or tissues, liquids or gases for specific purposes. Invasive devices, other than surgically invasive devices which are intended to administer medicinal products by inhalation, fall under the Rule 20
These types of devices should be considered separately from the non-contact devices of Rule 1 because they may be indirectly invasive. They channel or store substances that will eventually be administered to the body. Typically these devices are used in transfusion, infusion, extracorporeal circulation and delivery of anaesthetic gases and oxygen.
In some cases devices covered under this rule are very simple delivery devices that rely on gravity.
| Class | Rule 2 | Examples |
|---|---|---|
| IIa | All non-invasive devices intended for channeling or storing blood, body liquids, cells or tissues, liquids or gases for the purpose of eventual infusion, administration or introduction into the body are classified as class IIa: - if they may be connected 1 to a class IIa, class IIb or class III active device; or if they are intended for use for channeling or storing blood or other body liquids or for storing organs, parts of organs or body cells and tissues, | Devices intended to be used as channels in active drug delivery systems, e.g. tubing intended for use with an infusion pump Devices used for channelling gases, e.g. antistatic tubing for anaesthesia, anaesthesia breathing circuits Syringes for infusion pumps Devices intended to channel blood ( e.g. in transfusion, extracorporeal circulation) Devices intended for temporary storage and transport of organs for transplantation ( i.e . containers, bags) |
MDCG 2021- 24
| Devices intended for long term storage of biological substances and tissues such as corneas, sperm, human embryos, etc. ( i.e. containers, bags) Fridges/freezers specifically intended for storing blood, tissues etc. Tubings/blood lines for extracorporeal treatment (dialysis and apheresis therapies) | ||
|---|---|---|
| IIb | - except for blood bags; blood bags are classified as class IIb. | Blood bags without a substance which, if used separately, can be considered to be a medicinal product |
| I | In all other cases, such devices are classified as class I | Non-invasive devices that provide a simple channelling function, with gravity providing the force to transport the liquid, e.g. administration sets for infusion Devices intended to be used for a temporary containment or storage function , e.g. cups and spoons specifically intended for administering medicines 2 Empty syringes without needles |
Practical issues of classification
If a device, e.g. tubing, intended to be used for a purpose that would require it to be connected to an active device, such a device will be automatically in class IIa, unless the manufacturer clearly states that it should not be connected to an active device of class IIa or higher.
Note 1: 'May be connected to an active device' - such a connection is deemed to exist between a non-active device and an active device where the non-active device forms a link in the transfer of the substance between the patient and the active device and the safety and performance of one of the devices is influenced by the other device. For instance, this applies to tubing in an extracorporeal circulation system which is downstream from a blood pump and in the same blood flow circuit, but not directly connected to the pump.
Note 2: See also Chapter 3.1.6 regarding the possible measuring function.
Rule 3 Devices that modify biological or chemical composition of human tissues or cells, blood, other body liquids or other liquids intended for implantation or administration into the body
General explanation of the rule
These types of devices must be considered separately from the non-contact devices of Rule 1 because they are indirectly invasive. They modify substances that will eventually be administered into the body. This rule covers mostly the more sophisticated elements of extracorporeal circulation sets, dialysis systems and autotransfusion systems as well as devices for extracorporeal treatment of body fluids which may or may not be immediately reintroduced into the body, including cases where the patient is not in a closed loop with the device.
This rule covers also substances in direct contact with human cells, tissues or organs in vitro before their implantation or administration, without substances derived from human or animal origin such as human albumin.
| Class | Rule 3 | Examples |
|---|---|---|
| IIb | All non-invasive devices intended for modifying the biological or chemical composition of human tissues or cells, blood, other body liquids or other liquids intended for implantation or administration into the body are classified as class IIb, | Devices intended to remove undesirable substances out of the blood by exchange of solutes such as hemodialy s ers Devices intended to separate cells by physical means, e.g. gradient medium for sperm separation Haemodialysis concentrates Device removing specific blood cells (e.g. activated) by specific binding to a matrix |
| IIa | unless the treatment for which the device is used consists of filtration, centrifugation or exchanges of gas, heat, in which case they are classified as class IIa | Particulate filtration of blood in an extracorporeal circulation system. These are used to remove particles from the blood Centrifugation of blood to prepare it for transfusion or autotransfusion excluding centrifuges for manufacturing a medicinal product Removal of carbon dioxide from the blood and/or adding oxygen Warming or cooling the blood in an extracorporeal circulation system. |
| III | All non-invasive devices consisting of a substance or a mixture of substances intended to be used in vitro in direct contact with human cells, tissues or organs taken from the human body or used in vitro with human embryos before their implantation or administration into the body are classified as class III. | Substances or mixture of substances for transport, perfusion, storage of organs intended for transplantation that do not achieve the principal intended action by pharmacological, immunological or metabolic means |
MDCG 2021- 24
| IVF or ART products without principal pharmacological/metabolic action (substances or mixture of substances) IVF cell media without human albumin |
|---|
Practical issues of classification
These devices are normally used in conjunction with an active medical device covered under Rule 9 or Rule 11.
Filtration and centrifugation should be understood in the context of this rule as being done exclusively through mechanical methods.
Rule 4 - Devices that come into contact with injured skin or mucous membrane
General explanation of the rule
This rule applies to non-invasive devices as well as invasive devices that come into contact with injured skin or mucous membrane. This rule is intended to primarily cover wound dressings independently of the depth of the skin wound or mucosa wound. The conventional types of products , such as those used as a mechanical barrier , are well understood and do not result in any great hazard. There have also been rapid technological developments in this area, with the emergence of new types of wound dressings for which new types of claims are made, e.g. management of the microenvironment of a wound to enhance its natural healing mechanism.
More novel claims relate to the mechanism of healing by secondary intent, such as influencing the underlying mechanisms of granulation or epithelial formation or preventing contraction of the wound. Some devices used on breached dermis or breached mucosa may even have a lifesustaining or lifesaving purpose, e.g. when there is full thickness destruction of the skin or mucosa over a large area and/or systemic impact.
Dressings incorporating a substance which, if used separately, can be considered to be a medicinal product and that has an action ancillary to that of the dressing, fall within class III under Rule 14. Devices composed of other substances which are absorbed by or locally dispersed in the human body fall under Rule 21.
| Class | Rule 4 | Examples |
|---|---|---|
| I | All non-invasive devices which come into contact with injured skin or mucous membrane are classified as: - class I if they are intended to be used as a mechanical barrier, for compression or for absorption of exudates; | Wound dressings for skin or mucous, such as: absorbent pads, island dressings, cotton wool, wound strips, adhesive bandages (sticking plasters, band-aid) and gauze dressings which act as a barrier, maintain wound position or absorb exudates from the wound Ostomy bags |
| IIb | - class IIb if they are intended to be used principally for injuries to skin which have breached the dermis or mucous membrane and can only heal by secondary intent; | Are principally intended to be used with severe wounds: |
MDCG 2021- 24
| Dressings intended for ulcerated wounds having breached the dermis Dressings intended for burns having breached the dermis Dressings for severe decubitus wounds Dressings incorporating means of augmenting tissue and providing a temporary skin substitute | ||
|---|---|---|
| IIa | - class IIa if they are principally intended to manage the micro-environment of injured skin or mucous membrane; and | Hydrogel dressings for wounds or injuries that have not breached the dermis or can only heal by secondary intent Non-medicated impregnated gauze dressings Polymer film dressings |
| IIa | - class IIa in all other cases | |
| ! | This rule applies also to the invasive devices that come into contact with injured mucous membrane. | Dressings for nose bleeds (the purpose of the dressing is not to manage micro-environment) are in class I according to this rule Dental wound dressings not containing animal derived material |
Practical issues of classification
The classification of devices covered by this rule depends on the use intended by the manufacturer's, e.g. a polymeric film dressing would be in class IIa if the intended use is to manage the micro-environment of the wound or in class I if its intended use is limited to retaining an invasive cannula at the wound site. Consequently, it is impossible to say a priori that a particular type of dressing belongs to a given class without knowing its intended use as defined by the manufacturer. However, a claim that the device is interactive or active with respect to the wound healing process usually implies that the device is at least class IIa.
Most dressings that are intended for a use that falls under class IIa or IIb also perform functions that are in class I, e.g. that of a mechanical barrier. Such devices are nevertheless classified according to their intended use in the higher class.
For such devices incorporating a substance which, if used separately, can be considered to be medicinal product or a human blood derivative, or animal tissues or derivatives rendered non-viable, see Rule 14 or Rule 18 respectively.
Explanation of special concepts
Breached dermis or mucosa: the wound exposes at least partly the subcutaneous tissue.
Secondary intent: the wound heals by first being filled with granulation tissue; subsequently the epithelium grows back over the granulation tissue and the wound contracts. In contrast, primary intent implies that the edges of the wound are close enough or pulled together, e.g. by suturing, to allow the wound to heal before formation of granulation tissue.
Invasive devices
Rule 5 - Devices invasive with respect to body orifices
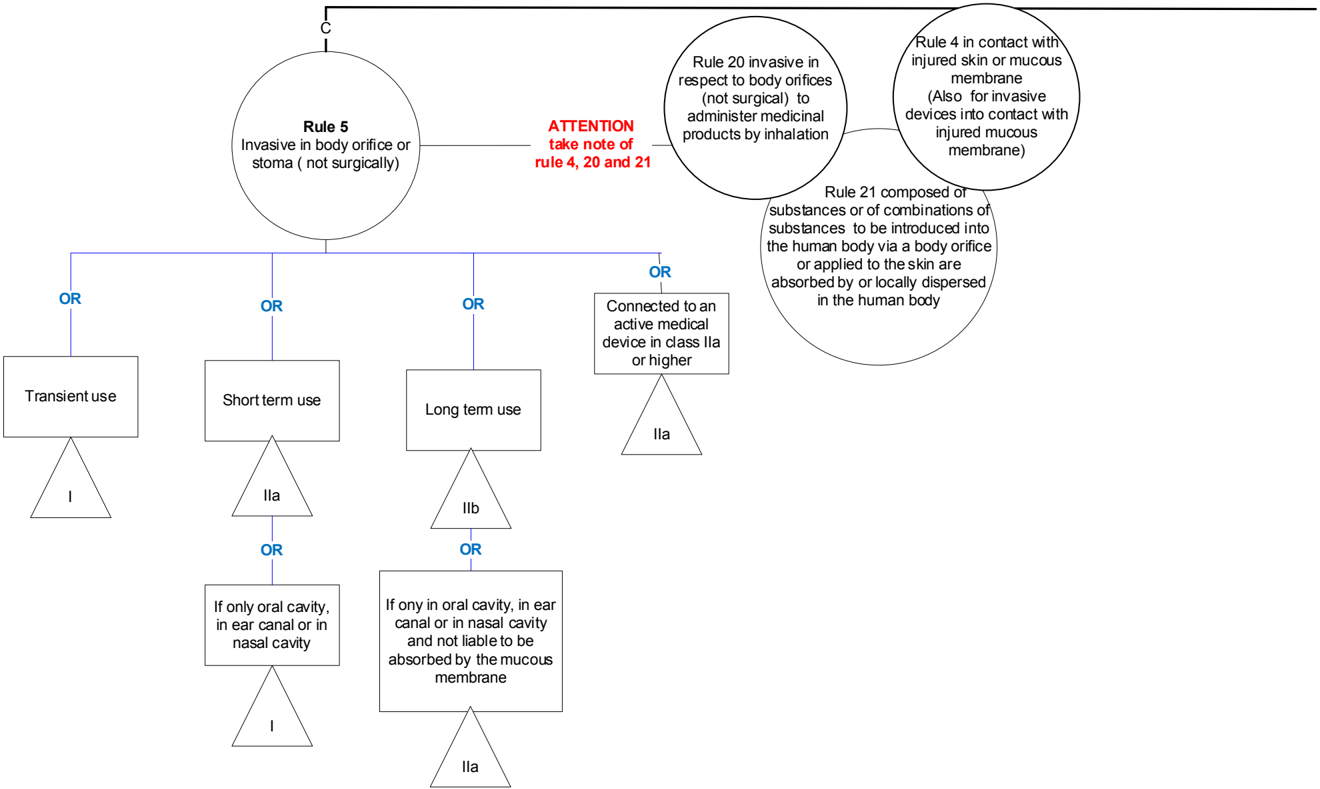
General explanation of the rule
Invasiveness with respect to body orifices must be considered separately from invasiveness that penetrates through a cut in the body surface (surgical invasiveness). For short term use, a further distinction must be made between invasiveness with respect to the less vulnerable anterior parts of the ear, mouth and nose and the other anatomical sites that can be accessed through natural body orifices. A surgically created stoma, which, for example, allows the evacuation of urine or faeces, is also to be considered a body orifice.
The devices covered by this rule tend to be diagnostic and therapeutic instruments used in particular specialties (otorhinolaryngology, ophthalmology, dentistry, proctology, urology and gynaecology).
| Class | Rule 5 | Examples |
|---|---|---|
| I | All invasive devices with respect to body orifices, other than surgically invasive devices, which are not intended for connection to an active device or which are intended for connection to a class I active device are classified as: class I if they are intended for transient use; | Handheld mirrors used in dentistry to aid in dental diagnosis and surgery Dental impression materials Stomach tubes Impression trays Examination gloves Urinary catheters intended for transient use Embryo transfer catheter and insemination catheter |
| IIa | - class IIa if they are intended for short-term use, | Short term corrective contact lenses Tracheal tubes Indwelling urinary catheters intended for short term use Gasses used for insufflation in the body Nasobilliary tubes |
| I | - except if they are used in the oral cavity as far as the pharynx, in an ear canal up to the ear drum or in the nasal cavity, in which case they are classified as class I; and | Materials for dental impressions Plastic syringe used to measure a quantity of medicinal product before oral administration to the patient Removable or fixed dental prostheses |
| IIb | - class IIb if they are intended for long-term use. | Urethral stents Long term corrective contact lenses Tracheal cannulae for tracheostoma for long term use Urinary catheters intended for long term use |
| IIa | - except if they are used in the oral cavity as far as the pharynx, in an ear canal up to the ear drum or in the nasal cavity and are not liable to be absorbed by the mucous membrane, in which case they are classified as class IIa. | Orthodontic wires Fixed dental prostheses Fissure sealants |
| IIa | All invasive devices with respect to body orifices, other than surgically invasive devices, intended for connection to a class IIa, class IIb or class III active device, are classified as class IIa | Tracheostomy or tracheal tubes connected to a ventilator Blood oxygen analysers placed under the eye-lid Powered nasal irrigators Fibre optics in endoscopes connected to surgical lasers Suction catheters or tubes for stomach drainage Dental aspirator tips Endoscopes using a light source in the visible spectrum |
|---|
Practical issues of classification
Regarding devices intended for connection to an active device: the strictest rule and sub-rule resulting in higher classification will apply. For instance a trachea cannula for long-term use need to be classified as class IIb.
Devices composed of substances which are absorbed by or locally dispersed in the human body may also fall under Rule 21
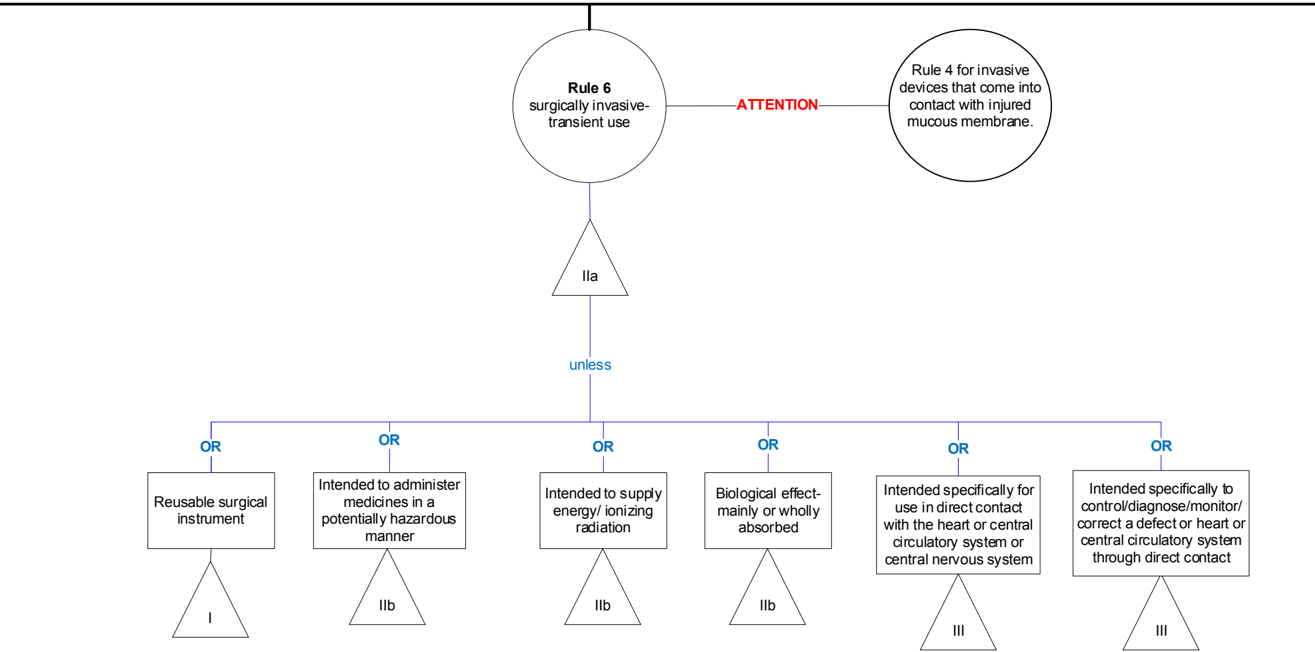
Rule 6 - Surgically invasive devices intended for transient use (<60 min)
General explanation of the rule
This rule primarily covers three major groups of devices: devices that are used to create a conduit through the skin (e.g. needles, cannulae), surgical instruments (e.g. scalpels, saws) and various types of catheters, suckers, etc.
| Class | Rule 6 | Examples |
|---|---|---|
| IIa | All surgically invasive 1 devices intended for transient use are classified as class IIa unless they: | Needles used for suturing Needles or syringes Lancets Single use scalpels and single use scalpel blades Surgical swabs Surgical gloves Swabs to sample exudates Guidewires or catheters used outside the central circulatory system |
| III | - are intended specifically to control, diagnose, monitor or correct a defect 2 of the heart or of the central circulatory system through direct contact with those parts of the body, in which case they are classified as class III 3 ; | Cardiovascular catheters (e.g. angioplasty balloon catheters, stent delivery catheters/systems), including related |
| Medical Device Coordination Group Document | Medical Device Coordination Group Document | MDCG 2021- 24 |
|---|---|---|
| guidewires, related introducers and dedicated 3 disposable cardiovascular surgical instruments e.g. electrophysiological catheters, electrodes for electrophysiological diagnosis and ablation, Catheters containing or incorporating sealed radioisotopes, where the radioactive isotope is not intended to be released into the body, if used in the central circulatory system Distal protection devices | ||
| I | - are reusable surgical instruments, in which case they are classified as class I; | Scalpels and scalpel handles Reamers Drill bits Saws, that are not intended for connection to an active device Retractors forceps, excavators and chisels Sternum retractors for transient use Staplers (outside the heart, central circulatory or central nervous system Dental Osteotomes |
| III | - are intended specifically for use in direct contact with the heart or central circulatory system or the central nervous system, in which case they are classified as class III; | Neuro-endoscopes Brain spatulas Direct stimulation cannulae Spinal cord retractors Spinal needles Cranium guide for use in craniotomy Dura mater protection; Bone punch for use on the cranium (Intended use: The dura mater protection is intended to protect the dura mater during surgical procedures. It has direct contact to the CNS. The bone punch can be used at the cranium. A direct contact to the CNS is possible during application.) Peripherally inserted central catheter (PICC) line Heart valve occluders, sizers and holders Cardiovascular drainage cannula specifically intended to circulate blood whilst located in the heart or central vascular system Cryo-ablation of the heart or spine Appliers/Forceps for aneurysm clips |
| IIb | - are intended to supply energy in the form of ionising radiation in which case they are classified as class IIb; - or | Catheters containing or incorporating sealed radioisotopes, where the radioactive isotope as such is not intended to be released into the body, excluding the central circulatory system |
|---|---|---|
| IIb | - have a biological effect or are wholly or mainly absorbed in which case they are classified as class IIb; | Viscoelastic solution for ophthalmic surgery |
| IIb | - are intended to administer medicinal products by means of a delivery system, if such administration of a medicinal product is done in a manner that is potentially hazardous 4 taking account of the mode of application, in which case they are classified as class IIb. | Refillable insulin pens Analgesia pumps |
Practical issues of classification/Explanation of special concepts
Note 1 : Terms such as 'surgically invasive device', 'central circulatory system', 'central nervous system' and 'reusable surgical instruments' are defined in Section 2 of Annex VIII to the MDR. In particular surgical instruments connected to an active device are not considered to be 'reusable surgical instruments'.
Note 2: The expression 'correct a defect' does not cover auxiliary devices that are used in heart surgery procedures, e.g. clamps, aortic punch instruments. The first indent of this rule does not apply to aortic punches and similar cutting instruments which perform a function similar to that of a scalpel.
Note 3: 'Dedicated' means that the intended purpose of the device or accessory is to specifically control, diagnose, monitor or correct a defect of the heart or of the central circulatory system.
Note 4: The concept of 'potentially hazardous manner' is related to the characteristics of the device and not to the competence of the user.
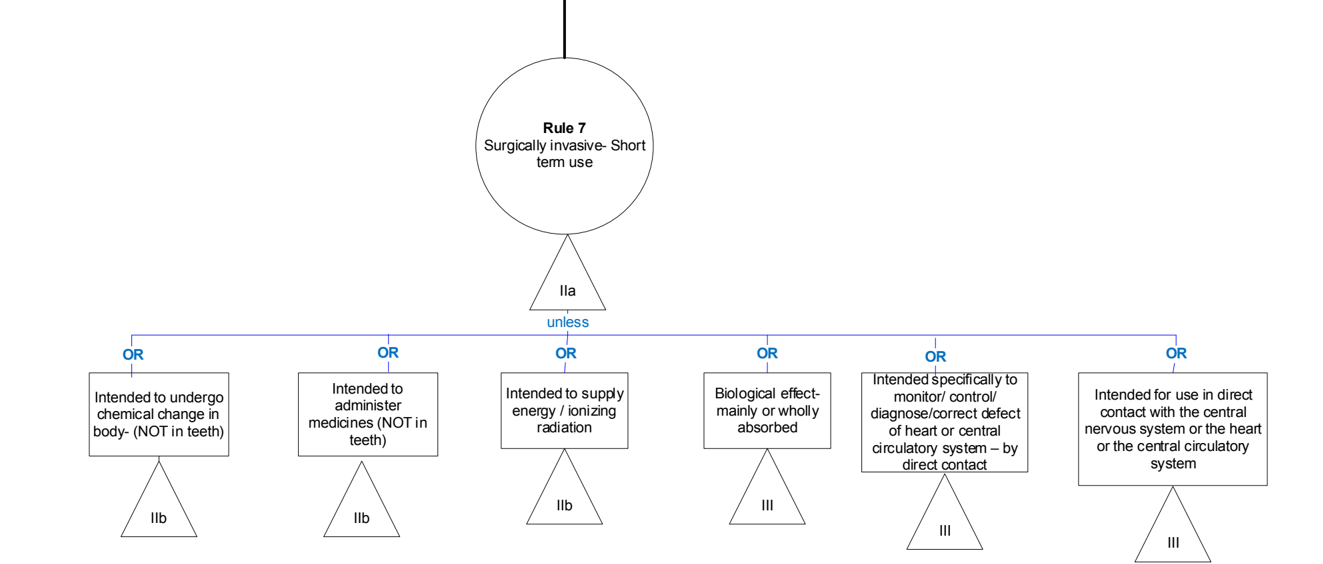
Rule 7 -Surgically invasive devices intended for short-term use (> 60 min <30 days)
General explanation of the rule
These are mostly devices used in the context of surgery or post-operative care (e.g. clamps, drains), infusion devices (e.g. cannulae, needles) and catheters of various types.
| Class | Rule 7 | Examples |
|---|---|---|
| IIa | All surgically invasive devices intended for short-term use are classified as class IIa unless they: | Clamps Infusion cannulae Skin closure devices Temporary filling materials |
| Arthroscopy trocars Insufflation gases for surgically invasive endoscopic procedures | ||
|---|---|---|
| III | are intended specifically to control, diagnose, monitor or correct a defect of the heart or of the central circulatory system through direct contact with those parts of the body, in which case they are classified as class III; | Cardiovascular catheters Cardiac output probes Temporary pacemaker leads Thoracic catheters intended to drain the heart, including the pericardium Carotid artery shunts Ablation catheter Heart bypass cannula (aortic perfusion cannula and venous drainage cannula) Peripherally inserted central catheter (PICC) line and central line |
| III | are intended specifically for use in direct contact with the heart or central circulatory system or the central nervous system, in which case they are classified as class III; | Neurological catheters Cortical electrodes Central venous/vascular catheters |
| IIb | are intended to supply energy in the form of ionizing radiation in which case they are classified as class IIb; | Brachytherapy devices |
| III | have a biological effect or are wholly or mainly absorbed in which case they are classified as class III; | Absorbable sutures |
| IIb | are intended to undergo chemical change in the body in which case they are classified as class IIb, except if the devices are placed in the teeth; or. | Vascular closure devices Haemostatic foams |
| IIb | are intended to administer medicines 1 , in which case they are classified as class IIb. | Temporal dialysis catheter, CVVH catheter |
Practical issues of classification
Note 1: Administration of medicinal products is more than just channelling, it implies also storage and/or control of the volume and rate of delivery. Implanted capsules for the slow release of medicines, placed on the market as a single integral product, are governed by Directive 2001/83/EC or Regulation (EC) No 726/2004.
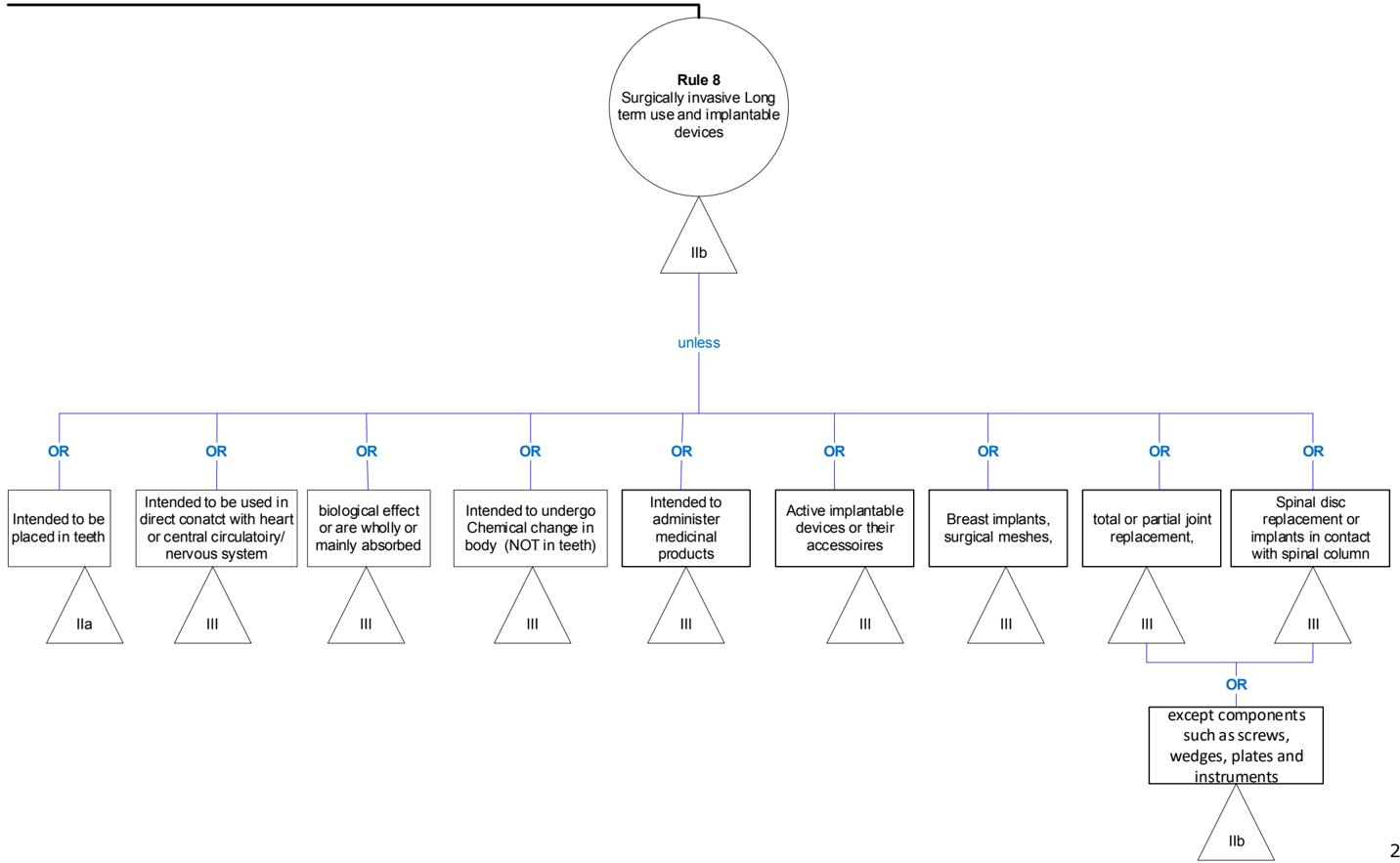
Rule 8 - Implantable devices and long-term surgically invasive devices (> 30 days)
General explanation of the rule
These are mostly implants in the orthopaedic, dental, ophthalmic and cardiovascular fields as well as soft tissue implants such as implants used in plastic surgery.
| Class | Rule 8 | Examples |
|---|---|---|
| IIb | All implantable devices and long-term surgically invasive devices are classified as class IIb unless they: | Artificial ligaments for reinforcement 2 . Dental implants and abutments Shunts Peripheral stents and peripheral valves Plates Intra-ocular lenses Internal closure devices (including vascular closure devices 1 ) Tissue augmentation implants (excluding breasts) Peripheral vascular catheters for long-term use Peripheral vascular grafts and stents Penile implants Non-absorbable sutures, non-biodegreable bone cements and maxillo-facial implants, visco- elastic surgical devices intended specifically for ophthalmic anterior segment surgery 2 Pedicle screws |
| IIa | - are intended to be placed in the teeth 3 , in which case they are classified as class IIa; | Bridges and crowns Dental filling materials and pins Dental alloys, ceramics and polymers |
| III | - are intended to be used in direct contact with the heart, the central circulatory system or the central nervous system, in which case they are classified as class III; | Prosthetic heart valves Aneurysm clips Vascular prosthesis and stents Central vascular catheters for long-term use Spinal stents CNS electrodes Cardiovascular sutures Permanent and retrievable vena cava filters Septal occlusion devices Intra-aortic balloon pumps External left ventricular assisting devices |
| III | - have a biological effect or are wholly or mainly absorbed, in which case they are classified as class III; | Long term absorbable sutures Adhesives and implantable devices claimed to be bioactive through the attachment of surface coatings such as phosphoryl choline Biodegrable Bone Cements Elastoviscus fluids for joint movement(eg. hyaluronan of non-animal origin) |
| III | - are intended to undergo chemical change 4 in the body in which case they are classified as class III, except if the devices are placed in the teeth |
MDCG 2021- 24
| III | - are intended to administer medicinal products, in which case they are classified as class III; | Rechargeable non-active drug delivery systems Peritoneal dialysis |
|---|---|---|
| III | - are active implantable devices or their accessories, in which cases they are classified as class III; | Cochlear implants and accessories Implantable cardiac pacemakers Implantable cardioverter defibrillators (ICD) Leads, electrodes, adaptors for pacemakers and implantable defibrillators Implantable nerve stimulators Implantable bladder stimulators Implantable sphincter stimulators Accessories to active implantable devices (with or without contact to the heart), be it implantable or non-implantable active or not 5 : torque wrench for implantable pulse generator / implantable cardioverter defibrillator cables for programmer / pacing system analyser magnet for Implantable Pulse Generator / Implantable Cardioverter Generator programmer or an external transmitter intended for activating or controlling the implantable part of the device implantable pacemaker leads |
| III | - are breast implants or surgical meshes, in which cases they are classified as class III; | Breast implants Breast tissue expanders Surgical meshes for hernia repair Tension free vaginal tape |
| III | - are total or partial joint replacements, in which case they are classified as class III, with the exception of ancillary components such as screws, wedges, plates and instruments; or | Hip, knee Shoulder Ankle |
| III | - are spinal disc replacement implants or are implantable devices that come into contact with the spinal column, in which case they are classified as class III with the exception of components such as screws, wedges, plates and instruments | Spinal disc replacement implants Spinal implants: hooks that fix the rod on the spinal column Stems that are implantable in contact with the spinal column Device placed in the disc space Interbody fusion devices |
Practical issues of classification
Note 1: For closure of arteriotomies in the peripheral vascular system (refer to definition of central circulatory system).
Note 2: These devices are implants because in normal conditions a significant amount of the substance remains at the surgical site after the procedure. Rule 18 may also be relevant.
Note 3: Implants without bioactive coatings intended to secure teeth or prostheses to the maxillary or mandibular bones are in Class IIb following the general rule.
Note 4 : The clause about chemical change under this rule does not apply to products such as bone cements where the chemical change takes place during the placement and does not continue in long term.
Note 5: Also non-implantable and non-active accessories to AIMDs should be classified as Class III under Rule 8.
Active devices
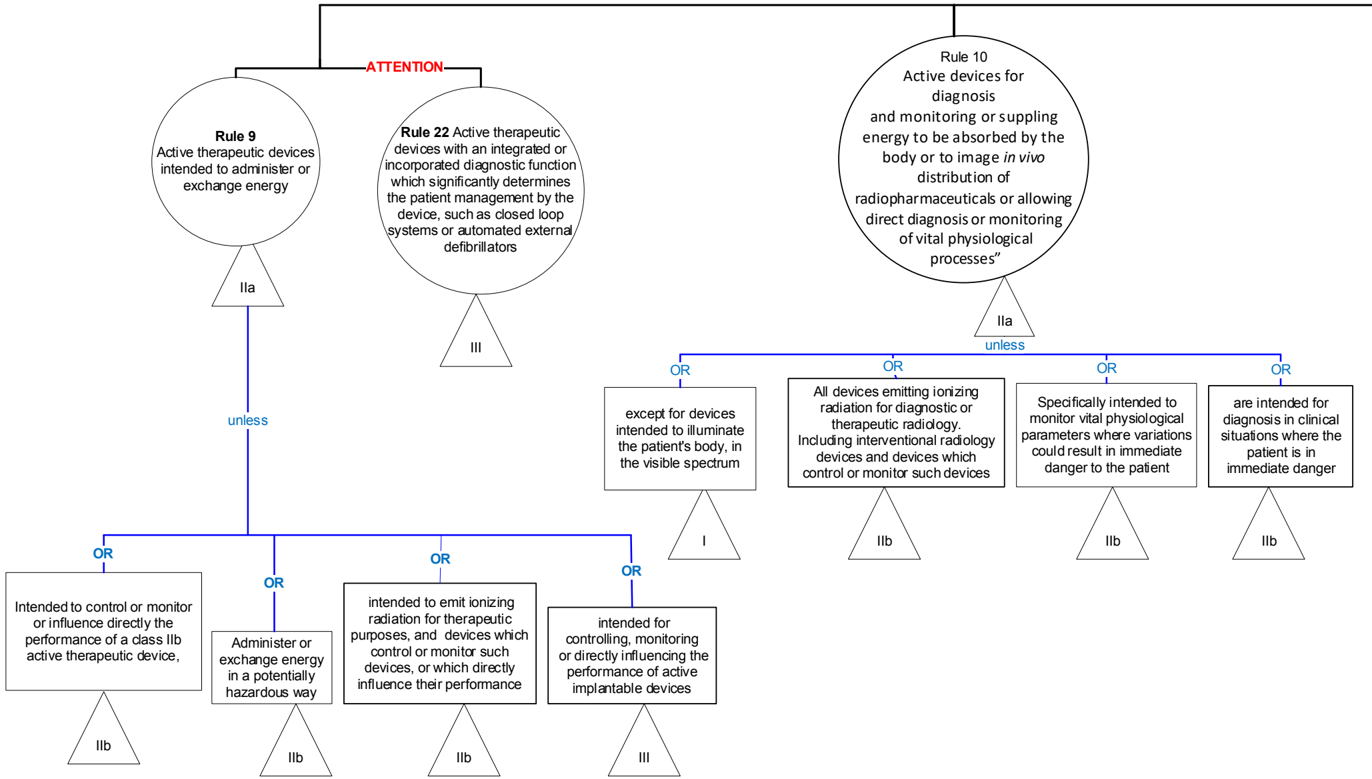
Rule 9 -Active therapeutic devices intended to administer or exchange energy 1 , as well as active devices intended to control/monitor/directly influence certain devices
General explanation of the rule
This rule covers many different groups of devices, such as:
- electrical equipment used in surgery such as lasers and surgical generators;
- stimulation devices;
- devices intended to emit ionizing radiation 2 for therapeutic purposes, including devices which control or monitor such devices, or which directly influence their performance;
- devices intended for controlling, monitoring or directly influencing the performance of active implantable devices.
Active implantable devices are covered by Rule 8.
Rule 22 may also apply to active therapeutic devices 27 .
| Class | Rule 9 | Examples |
|---|---|---|
| IIa | All active therapeutic devices intended to administer or exchange energy are classified as class IIa | Electrical and/or magnetic and electromagnetic energy: muscle stimulators external bone growth stimulators TENS devices eye electromagnets electrical acupuncture Thermal energy: heat exchangers, except the types described below Mechanical energy: powered dermatomes powered drills dental hand pieces Light: phototherapy for skin treatment and for neonatal care Sound: external hearing aids Ultrasound: equipment for physiotherapy Sleep apnoea ventilators without monitoring function |
| IIb | unless their characteristics are such that they may administer energy to or exchange energy with the human body in a potentially hazardous way, taking account of the nature, the density and site of application of the energy, in which case they are classified as class IIb. | Kinetic energy: lung ventilators Thermal energy: incubators for babies blood warmers electrically powered heat exchangers (with patients incapable of reacting, communicating /or who are without a sense of feeling) Electrical energy: high-frequency electrosurgical generators, and electrocautery equipment, including their electrodes external pacemakers and external defibrillators with no integrated or incorporated diagnostic function electroconvulsive therapy equipment Coherent light: |
MDCG 2021- 24
| surgical lasers Ultrasound: lithotriptors, surgical ultrasound devices high-intensity focused ultrasound (HIFU) | ||
|---|---|---|
| IIb | All active devices intended to control or monitor the performance of active therapeutic class IIb devices, or intended directly to influence the performance of such devices are classified as class IIb. | External feedback systems for active therapeutic devices |
| IIb | All active devices intended to emit ionizing radiation for therapeutic purposes, including devices which control or monitor such devices, or which directly influence their performance, are classified as class IIb | Brachytherapy therapy devices if the device also generates the radiation Therapeutic cyclotrons and linear accelerators Therapeutic X-ray sources |
| III | All active devices that are intended for controlling, monitoring or directly influencing the performance of active implantable devices are classified as class III | Programming units and pacing system analysers Cardioscopes with pacing pulse indicators specifically intended to monitor active implantable devices Programmer for: implantable Pulse Generator (IPG); implantable Cardioverter Defibrillator (ICD) implantable Loop Recorder Remote monitoring devices for active implantable devices |
Practical issues of classification
Note 1 : The decision as to whether a medical device administers or exchanges energy to and from the human body in a potentially hazardous way should take into account the following factors. The concept of 'potentially hazardous' is dependent on the type of technology involved and the intended application of the device to the patient and not on the measures adopted by the manufacturer in view of good design management ( e.g . use of technical standards, risk analysis). For instance, all devices intended to emit ionizing radiation, all lung ventilators and lithotriptors should be in Class IIb. However, the manufacturer's obligation to comply with design requirements and solutions adopted, exist independently from the classification system. Devices for illumination, i.e. administration of rays in visible light spectrum, for use in the process of diagnosis are classified as class IIa or higher, e.g. visual electrophysiology or fundoscopes for examination of the eye; whereas devices for illumination for purposes other than in the process of diagnosis, e.g. therapeutic purposes (light therapy), are class I.
Note 2 : 'Ionising radiation' means energy transferred in the form of particles or electromagnetic waves of a wavelength of 100 nanometres or less (a frequency of 3 × 10 15 hertz or more) capable of producing ions directly or indirectly (Directive 2013/59/EURATOM, Article 4, 46).
Rule 10 - Active devices for diagnosis and monitoring or intended for diagnostic or therapeutic radiology General explanation of the rule
This rule covers a whole range equipment in various fields for capture of physiological signals, as well as specifically therapeutic and diagnostic radiology. Note that devices for recording diagnostic X-ray images are covered by Rule 17. Devices specifically intended to monitor active implantable devices fall under Rule 8 or Rule 9.
| Class | Rule 10 | Examples |
|---|---|---|
| IIa | Active devices intended for diagnosis and monitoring are classified as class IIa: - if they are intended to supply energy which will be absorbed by the human body, | Magnetic resonance equipment Pulp testers Evoked response stimulators Diagnostic ultrasound |
| I | except for devices intended to illuminate 1 the patient's body, in the visible spectrum, in which case they are classified as class I; | Examination lamps Surgical microscopes intended to illuminate the patient's body in the visible spectrum Dermatoscopes with integrated light sources |
| IIa | - if they are intended to image in vivo distribution of radiopharmaceuticals; or | Gamma cameras Positron emission tomography and single photon emission computer tomography |
| IIa | - if they are intended to allow direct diagnosis 2 or monitoring of vital physiological processes 3 , | Electrocardiographs Electroencephalographs Electronic thermometers Electronic stethoscopes Electronic blood pressure measuring equipment |
| IIb | unless they are specifically intended for monitoring of vital physiological parameters 3 and the nature of variations of those parameters is such that it could result in immediate danger to the patient, for instance variations in cardiac performance, respiration, activity of the central nervous system, or they are intended for diagnosis in clinical situations where the patient is in immediate danger, in which cases they are classified as class IIb. | Blood gas analysers used in open heart surgery Apnoea monitors, including apnoea monitors in home care Patient monitors (intended use: Monitor intended for multi-parameter patient monitoring. The device will produce visual and audible alarms if any of the physiological parameters monitored vary beyond pre-set limits and timed alarm recordings will be produced.), for example in intensive care monitoring, e.g. blood pressure, temperature, oxygen saturation |
| IIb | Active devices intended to emit ionizing radiation 4 and intended for diagnostic or therapeutic radiology 5 , including interventional radiology devices 5 and devices which control or monitor 6 such devices, or which directly influence their performance, are classified as class IIb. | Diagnostic X-Ray machine Computed Tomography Devices |
Practical issues of classification / Explanation of special concepts
Note 1: 'Illuminate' means: to administer rays in visible light spectrum to for use in the process of diagnosis for example in visual electrophysiology and eye diseases, and not just to cast a light to improve the visibility. When performing diagnosis to, for example, perform visual electrophysiology in search of eye diseases, the intended purpose of the device is not limited to 'illumination', but may include diagnosis.
Note 2: A device is considered to allow direct diagnosis when it itself provides the diagnosis of the disease or condition in question or when it provides decisive information for the diagnosis (MDR 2017/745, Annex VIII, 3.7). For the definition of diagnosis see the MDCG Guidance document covering the borderline between medical devices and medicinal products.
Note 3: Vital physiological processes and parameters include, for example, respiration, heart rate, cerebral functions, blood gases, blood pressure and body temperature. Medical devices intended to be used for continuous surveillance of vital physiological parameters in anesthesia, intensive care or emergency care are in class IIb, whilst medical devices intended to be used to obtain readings of vital physiological signals as part of routine checkups or self-monitoring are in class IIa. A thermal imaging device intended to monitor blood flow is not considered to be a body temperature measuring device.
Note 4 : 'Ionising radiation' means energy transferred in the form of particles or electromagnetic waves of a wavelength of 100 nanometers or less (a frequency of 3 × 10 15 hertz or more) capable of producing ions directly or indirectly (Directive 2013/59/EURATOM, Article 4, 46).
Note 5 : Therapeutic interventional radiology refers to diagnosis being carried out during a surgical procedure.
Note 6 : This refers to active devices for the control, monitoring or influencing the emission of ionizing radiation and not to the subsequent processing, recording or viewing of the resulting image. Devices for recording diagnostic X-ray images are covered by Rule 17.
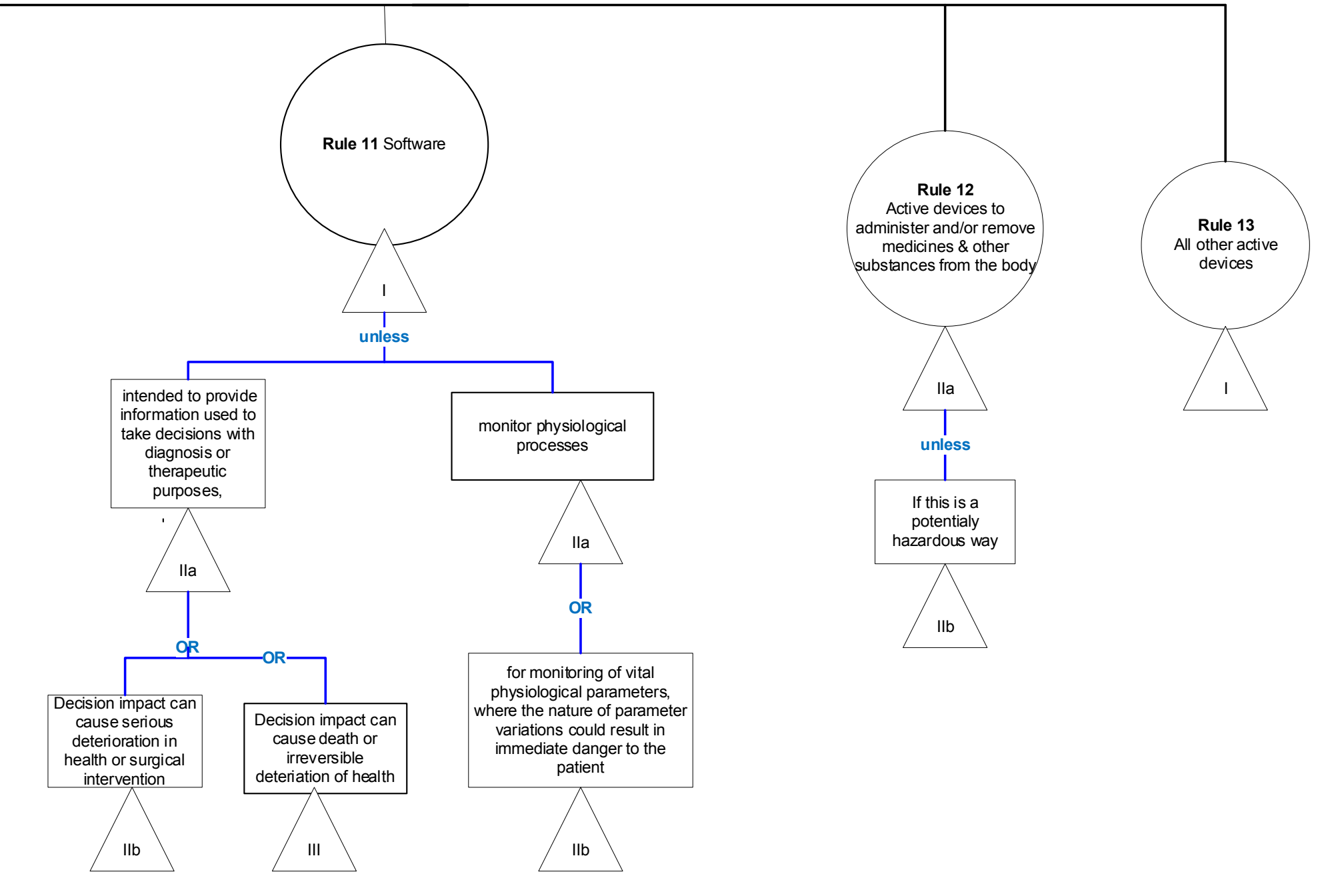
Rule 11 - Software intended to provide information to inform decisions with diagnosis or therapeutic purposes or software intended to monitor physiological processes.
General explanation of the rule
Rule 11 describes and categorizes the risk of software based on the combination of the significance of the information provided by the software to the healthcare decision and the healthcare situation or patient's condition.
This rule also distinguishes between MDSW (medical device software) intended to monitor vital and non-vital physiological processes (the subrule only applies to software intended for monitoring purposes only).
MDCG 2021- 24
Software or equipment attached to devices which do not specifically enable device(s) to be used in accordance with their intended purpose, or to specifically or directly assist the medical functionality of the device in terms of its/their intended purpose do not meet the definition of an accessory.
Software used in conjunction with medical devices(s) which solely record, store or display information would generally not be considered devices (see guidance MDCG 2019-11, section 3.3 for further detail). For example, software analogous to diaries for recording insulin doses would not be considered devices, unless an analysis is performed on the data or the device in some way alters the patients treatment, prescription, doses etc.
| Class | Rule 11 | Examples |
|---|---|---|
| IIa | Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause: | MDSW intended to rank therapeutic suggestions for a health care professional based on patient history, imaging test results, and patient characteristics, for example, MDSW that lists and ranks all available chemotherapy options for BRCA-positive individuals. Cognitive therapy MDSW where a specialist determines the necessary cognitive therapy based on the outcome provided by the MDSW. |
| III | - death or an irreversible deterioration of a person's state of health 1 , in which case it is in class III; or | MDSW intended to perform diagnosis by means of image analysis for making treatment decisions in patients with acute stroke. |
| IIb | -aserious deterioration of a person's state of health 1 or a surgical intervention, in which case it is classified as class IIb. | A mobile app intended to analyse a user's heartbeat, detect abnormalities and inform a physician accordingly. MDSWintended for diagnosing depression based on a score resulting from inputted data on patient symptoms (e.g. anxiety, sleep patterns, stress etc.). |
| IIa | Software intended to monitor physiological processes is classified as class IIa, | MDSW intended to monitor physiological processes that are not considered to be vital. Devices intended to be used to obtain readings of vital physiological signals in routine check-ups including monitoring at home. |
| IIb | except if it is intended for monitoring of vital physiological parameters 3 , where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb. | Medical devices including MDSW intended to be used for continuous surveillance of vital physiological processes in anaesthesia, intensive care or emergency care. |
| I | All other software is classified as class I. | MDSW app intended to support conception by calculating the user's fertility status based on a validated statistical algorithm. The user inputs health data including basal body temperature |
| (BBT) and menstruation days to track and predict ovulation. The fertility status of the current day is reflected by one of three indicator lights: red (fertile), green (infertile) or yellow (learning phase/cycle fluctuation). |
|---|
Practical issues of classification
Note 1 : The MDR contains several references to 'serious deterioration of a person's state of health' and to 'surgical intervention', notably in the vigilance or clinical investigation context. Further horizontal guidance may be provided in the future and will be available at: https://ec.europa.eu/health/md\_sector/new\_regulations/guidance\_en .
Note 2: For the classification of software, it is needed to consider the intended purpose, intended population (including e.g. diseases to be treated and/or diagnosed), context of use ( e.g. intensive care, emergency care, home use) of the software and of the information provided by the software as well as of the possible decisions to be taken.
Note 3 Medical device software should be classified in the same way, regardless of the software's location or the type of interconnection between the software and a (hardware) device. However, in line with implementing rule 3.3 Annex VIII to the MDR, software which drives a device or influences the use of a device shall fall within the same class as the device.
For further information on qualification and classification of software consult: MDCG 2019-11 Qualification and classification of software Regulation (EU) 2017/745 and Regulation (EU) 2017/746 28 .
Rule 12 - Active devices intended to administer and/or remove medicinal products, body liquids or other substances to or from the body
General explanation of the rule
This rule is intended to primarily cover drug delivery systems and anaesthesia equipment. If the device's intended route of drug delivery is pulmonary, Rule 20 applies.
| Class | Rule 12 | Examples |
|---|---|---|
| IIa | All active devices intended to administer and/or remove medicinal products, body liquids or other substances to or from the body are classified as class IIa, | Suction pump Feeding pumps Jet injectors for vaccination |
| Elastomeric pumps or balloon pumps for infusion | ||
|---|---|---|
| IIb | unless this is done in a manner that is potentially hazardous, taking account of the nature the substances involved, of the part of the body concerned and of the mode of application which case they are classified as class IIb. | of in Infusion pumps Ventilators Anaesthesia machines Anaesthetic vaporisers Dialysis equipment Blood pumps for heart-lung machines Hyperbaric chambers Pressure regulators for medical gases Medical gas mixers Moisture exchangers in breathing circuits if used on unconscious or non-spontaneously breathing patients Oxygen concentrator used to deliver oxygen enriched air directly to the patient |
Rule 13 - All other active devices
General explanation of the rule
This is a fallback rule to cover all active devices not covered by other rules.
| Class | Rule 13 | Examples |
|---|---|---|
| I | All other active devices are classified as class I. | Electric wheelchairs Dental curing lights Electric hospital beds Patient hoists Dental patient chairs |
Special rules
Rule 14 - Devices incorporating, as an integral part, an ancillary medicinal product, and medicinal products derived from human blood or blood plasma
General explanation of the rule
This rule covers those devices incorporating, as an integral part, a substance which, if used separately, can be considered to be a medicinal product, with an action ancillary to that of the device. The principal intended action of the device must not be achieved through a pharmacological, immunological or metabolic action of the incorporated medicinal substance 1 . If the principal intended action of the device is mainly achieved by the action of the medicinal substance, the integral product shall be governed by Directive 2001/83/EC or Regulation (EC) No 726/2004 of the European Parliament and of the Council, as applicable. For more information see the MDCG Guidance document covering the borderline between medical devices and medicinal products. The same principle applies if a substance of animal origin, as defined in Art. 2(17), is part of the device and has an action ancillary to that of a device.
| Class | Rule 14 | examples |
|---|---|---|
| III | All devices incorporating, as an integral part, a substance which, if used separately, can be considered to be a medicinal product, as defined in point 2 of Article 1 of Directive 2001/83/EC, including a medicinal product derived from human blood or human plasma, as defined in point 10 of Article 1 of that Directive, and that has an action ancillary to that of the devices, are classified as class III. | Bone cement with antibiotics Condoms with spermicide Catheters coated with anticoagulants (e. g. heparin) Endodontic materials with antibiotics Ophthalmic irrigation solutions principally intended for irrigation, which contain components supporting the metabolism of the endothelial cells of the cornea Dressings incorporating an antimicrobial agent where the agent has an ancillary action on the wound Drug eluting stents (e.g. coronary, pulmonary) Surgical sealants containing human serum albumin or thrombin Implants coated with human fibrinogen Blood bags incorporating heparin or other substances as anticoagulant agents which, if used separately, can be considered to be a medicinal product IVF cell media with human albumin 2 Intra Uterine Devices (IUD) containing medicinal substances 3 including copper or silver Catheter lubrication gels containing analgesia e.g. lidocaine |
Practical issues of classification
Note 1: The definition of 'substance' in Article 1 (3) of Directive 2001/83/EC is applicable as far as not excluded by the MDR (e. g. exclusion criteria Article 1 (6) (h)).
Note 2: IVF cell media with human albumin are in class III according to Rule 14 and Rule 3. (Rule 14 applies, being the strictest, according to MDR, Annex VIII, chapter II, point 3.5.)
Note 3: This only applies if the effect of the medicinal substance is ancillary - if it is the main effect, the IUD would be classified as a medicinal product
N.B.: For the clarification of the meaning and application of 'a substance which, if used separately, can be considered to be a medicinal product' and of 'has an action ancillary to that of the device', clarification which is relevant for the application of this rule, see clarification provided in the respective section of the MDCG Guidance document covering the borderline between medical devices and medicinal products.
Rule 15 - Devices used for contraception or prevention of sexually transmitted diseases
General explanation of the rule
This rule covers invasive, implantable and non-invasive medical devices for contraception or prevention of sexually transmitted diseases, i.e. this rule is not limited to devices based on physical barriers. The rule covers contraception devices and devices used in prevention of transmission of sexually transmitted diseases, where non-invasive, devices for transient or short term use are of class IIb and long-term use or implantable devices are of class III.
Devices for fertility monitoring or testing to facilitate conception (and not used in contraception) are not covered by this rule.
Some devices may have dual functions, intended to be used both for contraception and prevention of the transmission of sexually transmitted diseases, e.g. condoms.
| Class | Rule 15 | Examples |
|---|---|---|
| IIb | All devices used for contraception or prevention of the transmission of sexually transmitted diseases are classified as class IIb, | Condoms and femidoms (internal condoms) Contraceptive diaphragms Fertility monitors and medical device software intended to be used in contraception (e.g. by using the basal body temperature) |
| III | unless they are implantable or long term invasive devices, in which case they are classified as class III. | Tubal ligation devices (e.g. clips or rings) Non-hormonal intrauterine contraceptive devices (IUCD or ICD) |
Rule 16 - Specifically disinfecting, cleaning, rinsing, hydrating or sterilising devices
General explanation of the rule
This rule is intended to cover various products used specifically with contact lenses such as solutions intended for storing contact lenses and solutions used to support contact lenses placed on the ocular surface.
The rule also covers substances and equipment specifically intended for the disinfection or sterilisation of devices intended by the manufacturer to be sterilised or disinfected prior to use, as referred to in MDR Article 2(1).
This rule does not apply to physical means for the cleaning of medical devices, such as ultrasound and brushes for general use. Such products will only be medical devices if they are specifically intended for the cleaning, disinfection or sterilisation of medical devices. Devices specifically intended for physical cleaning of contact lenses are covered by this rule.
| Class | Rule 16 | Examples |
|---|---|---|
| IIb | All devices intended specifically to be used for disinfecting, cleaning, rinsing or, where appropriate, hydrating contact lenses are classified as class IIb. | Contact lens storing solutions Cleaners for contact lenses Ultraviolet, vibration, or ultrasonic devices for cleaning and disinfecting contact lenses |
| IIa | All devices intended specifically to be used for disinfecting or sterilising medical devices are classified as class IIa, | Disinfecting solutions specifically intended for non-invasive medical devices Washer-disinfectors intended specifically for disinfecting non-invasive medical devices Sterilisers intended to sterilise medical devices in a medical environment |
| IIb | unless they are disinfecting solutions or washer-disinfectors intended specifically to be used for disinfecting invasive devices, as the end point of processing 1 , in which case they are classified as class IIb. | Solutions/disinfectors for trans oesophageal ultrasound probes) Washer-disinfector equipment specifically for disinfecting endoscopes or other invasive devices at the end point of processing (e. g. dental equipment) Disinfectants for the fluid pathways of haemodialysis equipment Denture disinfecting products |
| ! | This rule does not apply to devices that are intended to clean devices other than contact lenses by means of physical action only. | Brushes specifically intended to clean medical devices by mechanical action Ultrasonic devices (for other devices than contact lenses) |
Practical issues of classification
Note 1: Including disinfecting solutions or washer-disinfectors that require no further processing for a disinfected device ready to be used. The rule also covers devices where the intended use is to disinfect prion contaminations.
Rule 17 - Devices to record X-ray diagnostic images
General explanation of the rule
This rule covers stand-alone X-ray detectors and sensors as recording devices used in several types or modalities of medical imaging procedures, each of which uses different technologies and techniques. It covers non-active devices and active devices used to record X-ray diagnostic images of the human body. The intention of the rule is to cover primarily digital devices and analogous recording media, but not media (including digital media) used for subsequent image processing and storage.
| Class | Rule 17 | Examples |
|---|---|---|
| IIa | Devices specifically intended for recording of diagnostic images generated by X-ray radiation are classified as class IIa. | Digital x-ray detectors for recording images Photostimulable phosphor plates X-ray films |
Practical issues of classification
Note 1: Devices intended to emit ionizing radiation for diagnostic and or therapeutic purposes are not covered by this rule. See Rule 10.
Rule 18 - Devices manufactured utilizing tissue or cells of human or animal origin or their derivatives
General explanation of the rule
This rule covers devices manufactured utilizing tissues or cells of human or animal origin, or their derivatives, which are non-viable or rendered non-viable, i.e. where there is no longer any capacity for cellular metabolic activity. This includes devices containing derivatives of human origin that have an ancillary action to that of the device, as well as devices that contain or are made of animal tissues (non-derivative) that have been rendered non-viable, or their derivatives.
| Class | Rule 18 | Examples |
|---|---|---|
| III | All devices manufactured utilising tissues or cells of human or animal origin, or their derivatives 1 , which are non- viable or rendered non-viable, are classified as class III, | Animal derived biological heart valves Porcine xenograft dressings |
MDCG 2021- 24
| Devices made from animal sourced collagen/gelatine Devices utilising hyaluronic acid of animal origin Substance-based devices containing collagen for use in body orifices Collagen dermal fillers Bone graft substitutes | ||
|---|---|---|
| I 3 | unless such devices are manufactured utilising tissues or cells of animal origin, or their derivatives, which are non-viable or rendered non-viable and are devices intended to come into contact with intact skin only. | Leather components of orthopaedic appliances |
Practical issues of classification
Note 1 : Derivatives are products that are processed from animal tissues and excludes products made by animals e.g. milk, silk, beeswax, honey, propolis, royal jelly, hair, lanolin.
Note 2 : The industrial manufacturing process for some devices may employ raw materials which contain small amounts of tallow or tallow derivatives (e.g. stearates in polymers) for example for greasing the moulds. Such substances, which may be present in the final device only as trace amounts, are not considered as derivatives of animal tissues for the purpose of this rule which therefore does not apply. However, if such substances are a constituent part of a device the rule will apply, for example wound dressings impregnated with tallow. Such an exemption applies only to classification, and not to other obligations related to devices manufactured utilising non-viable substances of animal origin, such as those referred to in Annex I 13.2.
Note 3 : This rule does not apply to devices manufactured utilizing tissues or cells of animal origin or their derivatives coming into contact with intact skin only. In such cases they are in class I in accordance to Rule 1. Intact skin includes the skin around an established stoma unless the skin is breached.
Rule 19 - Devices incorporating or consisting of nanomaterial 29
General explanation of the rule
The concept of internal exposure is a key element for the classification incorporating or consisting of nanomaterials. In its 'Opinion on the Guidance on the Determination of Potential Health Effects of Nanomaterials Used in Medical Devices' 30 , the Scientific Committee on Emerging and Newly Identified Health Risks (SCENIHR) states as a conclusion, that the potential risk from the use of nanomaterials in medical devices is
MDCG 2021- 24 mainly associated with the possibility for release of free nanoparticles from the device and the duration of exposure. Table 3 of the SCENIHR Opinion estimates both external and internal exposure based on the type of device, type of application, type (location) of contact, and duration of contact. As indicated in the explanation of table 3 of the SCENIHR opinion, the 'potential internal systemic exposure of all organ systems' is what is listed, which can be expected to occur after release of free nanoparticles from invasive devices as well as from non-invasive devices in contact with a breached or compromised body surface. This Table 3 uses the terms high/medium/low/negligible, as also used in the classification rule. This Table 3 could be used as a starting point for correct classification. This Table 3 indicates a value for potential contact and/or external exposure to the nanomaterial and potential internal systemic exposure of all organ systems However, every individual device needs to be classified taking into account its own specific characteristics with regard to the potential release of free nanoparticles taking also into account the exposure by the same nanomaterial via daily exposure routes., Also factors such as the number of nanomaterials in or on the product and the amount of product applied in the intended use have been taken into account. This Table 3 indicates a value for potential contact and/or external exposure to the nanomaterial and potential internal systemic exposure of all organ systems.
| Class | Rule19 | Examples |
|---|---|---|
| All devices incorporating or consisting of nanomaterial are classified as | ||
| III | -class III if they present a high 1 or medium potential for internal exposure 2 | Bone fillers with nanomaterials in their formulation (not polymerized before blood/tissue contact, and degradable) Superparamagnetic iron oxide nanoparticles (Intended use: thermal ablation of tumors or thermal modulation of the tumor microenvironment by submission to alternating magnetic fields) Intravascular catheter made of non-degradable polymer, with nano-coating 31 |
| IIb | - class IIb if they present a low 1 potential for internal exposure | Bone fixation screws/plates with a strongly bound nano-coating high potential Solution administration set made of non-degradable polymer, with a strongly bound nano-coating |
| IIa | - class IIa if they present a negligible 1 potential for internal exposure | Intravascular catheter for short term use made of non- degradable polymer, with nanomaterial embedded in the polymer matrix Solution administration set made of non-degradable polymer, with nanomaterial embedded in the polymer matrix Dental filling materials |
Practical issues of classification
Note 1: The high, medium, low or negligible potential of internal exposure is based on the combination of different factors such as the application site of a medical device, type of contact (e.g. tissue, cells or body fluids), contact time and the type of incorporation of the nanomaterial(s)
(free, fixed, embedded). When the nanomaterial is applied as coating on the surface of the device, it is important to consider the type of interaction with the material (chemisorption versus physisorption). When the nanomaterial is embedded in a matrix it will be important to consider the degradability of the material.
Note 2: Internal exposure: exposure may occur via injured skin or mucous membrane, (surgically) invasive devices and implantable devices.
Note 3: Dental materials which are placed in the patient's teeth in a paste form, where they are cured to a solid form, may release nanomaterials during a very short exposure time. For most of the exposure time, these devices contain firmly bound nanomaterials. In many cases, grinding and/or polishing takes place during the application of the device, and could also lead to exposure to nanomaterials. Such nanomaterials do not necessarily contain the original nanomaterials present in the paste formulation. It is very important to include this aspect in the risk assessment of such devices. For classification purposes the potential internal exposure to nanomaterials from these devices can generally be considered negligible. Classification of this kind of materials should be based on the first state, in this example, the short exposure to the paste form which has higher potential for release than the cured material.
Note 4: Devices with components incorporating nanomaterials that have no intended direct or indirect contact with users or patients, such as the tires of wheelchairs or walking frames made from rubber reinforced with carbon black nanomaterials, should be exempt from classification under Rule 19.
Note 5: Medical devices not incorporating or consisting of nanomaterials can still present a potential for internal exposure to nanomaterials due to degradation or wear processes. While it is very important to include this aspect in the risk assessment of such devices, it is not a factor to be considered when deciding the classification under Rule 19 since this rule is only applicable for medical devices incorporating or consisting of nanomaterials.
Rule 20 - Invasive devices, intended to administer medicinal product by inhalation
General explanation of the rule
This rule covers active and non-active medical devices with a respiratory route of drug delivery.
In contrast to other rules covering devices that administer medicinal products, Rule 20 is also specifically intended to cover medical devices where the impact of the medical device on the efficacy and safety of the administered medicinal product is critical. The rule also covers drugdelivery products that are intended to treat life-threatening conditions.
| Class | Rule 20 | Examples |
|---|---|---|
| IIa | All invasive devices with respect to body orifices, other than surgically invasive devices, which are intended to administer medicinal products by inhalation are classified as class IIa, | Spacer intended for metered dose inhalers (attached to the inhaler) unless treating life- threatening conditions. |
MDCG 2021- 24
| Inhalers for nicotine replacement therapy (nicotine not included) Oxygen delivery system with a nasal cannula unless treating life-threatening conditions Inhalers and nebulisers in case their mode of action has probably no essential impact on the efficacy and safety of the administered medicinal product or which are not intended to treat life-threatening conditions | ||
|---|---|---|
| IIb | unless their mode of action has an essential impact 1 on the efficacy and safety of the administered medicinal product or they are intended to treat life- threatening conditions, in which case they are classified as class IIb | Nebulisers (not pre-charged with a specific medicinal product) where the failure to deliver the appropriate dosage characteristics could be hazardous Spacer intended for metered dose inhalers attached to the inhaler. |
Practical issues of classification
Note 1. 'Essential impact' includes drug delivery systems where the device has a significant impact on factors that influence inhaled medicinal product deposition within the airways including inhalation flow, aerosol velocity, the particle size of the inhaled drug and the amount of drug reaching the patient.
Rule 21 Devices composed of substances that are introduced via a body orifice or applied to the skin
General explanation of the rule
This rule covers a wide range of exclusively substance-based medical devices. In this context, 'substance' means any matter that is part of the medical device, including those according to the definition of 'substance' in Article 1(3) of Directive 2001/83/EC. This is provided that they are not excluded by the MDR ( e.g. exclusion criteria in article 1 (6) (h)). The specific medical purpose is specified by the manufacturer from those listed in the indents of Article 2(1) MDR.
Importantly, if the principal intended action of such substance-based medical devices is achieved or supported by pharmacological, immunological or metabolic means, Directive 2001/83/EC or Regulation (EC) No 726/2004 or Rule 14 has to be applied to the product, respectively.
The classification takes into account the site of application of the medical device as well as the site where the medical device performs its action in or on the human body. For the purpose of this rule nails are also considered as falling under 'skin'.
Manufacturers of substance-based devices should provide clear information supporting the mode of action through which the substance achieves the intended specific medical purpose as a basis for the application of this rule, including the site of application as well as the site where the action is achieved in or on the body.
| Class | Rule 21 | Examples |
|---|---|---|
| III | Devices that are composed of substances or of combinations of substances that are intended to be introduced into the human body via a body orifice or applied to the skin and that are absorbed by or locally dispersed in the human body are classified as: - class III if they, or their products of metabolism, are systemically absorbed by the human body in order to achieve the intended purpose; | |
| III | - class III if they achieve their intended purpose in the stomach or lower gastrointestinal tract and they, or their products of metabolism, are systemically absorbed by the human body; | Na/Mg alginate, xyloglucan Fat absorbers that are systemically absorbed, themselves or their metabolites |
| IIa | - class IIa if they are applied to the skin or if they are applied in the nasal or oral cavity as far as the pharynx 1 , and achieve their intended purpose on those cavities; and | Substance-based formulations for skin treatment Salt water used e.g. as nose or throat sprays Oral cough treatments achieving their intended purpose in the oral cavity as far as the pharynx |
| IIb | - class IIb in all other cases. | Simethicone preparations for oral administration Active coal for oral administration Gel for vaginal moisturizing / vaginal lubricants Eye drops for hydration Ear drops 1, 2 Medical devices, for oral administration, for the treatment of diarrhoea, e.g. kaolin, diosmectite Medical devices, for oral administration, for the treatment of obesity, e.g. fructooligosaccharides, glucomannan |
Practical issues of classification
Products acting in the nasal or oral cavity may be to some extent be ingested or inhaled. These products will be class IIa devices if the products achieve their intended purpose solely in these cavities, and not in the respiratory tract, stomach or lower gastrointestinal tract.
N.B.: examples provided do not imply that the products are a priori qualified as devices. Classification rules apply after the qualification of the device has been established.
Note 1: There is a normal anatomical connection, the Eustachian tube, from the middle ear to the nasopharynx. If the Eustachian tube was to be accessed from the nasopharynx direction, this would be considered beyond the pharynx, so this does not fulfil the criteria of 'as far as the pharynx'.
Note 2: In the majority of cases the drops would only enter the ear as far as the ear drum. This is regarded as applied to the skin. The outer layer of the tympanic membrane is epithelium, so if there is an intact tympanic membrane, then the drops are applied only to the skin and has its action locally, and consequently the device would be class IIa. This will be the case unless the ear drum (tympanic membrane) is perforated and the product is intended to be used on perforated ear drums.
Rule 22 Active therapeutic devices, with an incorporated diagnostic function
General explanation of the rule
This rule is intended for therapeutic devices whose intended functionality is dependent to a significant degree on an integrated or incorporated diagnostic function.
Automated or 'closed-loop' therapeutic systems are systems in which relevant biological conditions are automatically monitored (uses feedback from physiological sensors) and is used to adjust a therapy in order to maintain or achieve a particular physiological state. Such devices are normally used in precision medicine and/or personalised therapies for obtaining optimal therapeutic efficacy. This rule covers systems such as autonomic pharmacological (drug-delivery) and neuromodulation systems.
| Class | Rule 22 | Examples |
|---|---|---|
| III | Active therapeutic devices with an integrated or incorporated diagnostic function 1 which significantly determines the patient management by the device, such as closed loop systems or automated external defibrillators, are classified as class III. | Automated external defibrillators (AED) including their pads/electrodes Semiautomatic external defibrillators Automated closed loop insulin delivery system Automated external infusion pumps with integrated sensors to adapt the infusion therapy Devices in brain-computer interfaces (BCIs) - used for e.g. motor control in severely paralyzed patients Closed-loop systems for deep brain stimulation (DBS) treatment of various neurological conditions |
| Closed-loop dynamic neurochemical control of therapeutic interventions e.g. target-controlled anaesthesia / infusion systems |
|---|
Practical issues of classification
Note 1 . 'Integrated or incorporated diagnostic function' means the functionality of a system including a physiological sensors e.g. the AED electrodes/pads using a feedback control to process and record changes in the patient's physiological state to continuously adjust a therapy. The diagnostic function can be physically integrated or a component of an external sub-system.
Footnotes
- Annex II 1.1 (f) MDR
- MDCG 2019-3
- MDCG 2019-9
- MDR Annex VIII 3.6
- MDR Annex VIII 2.8
- MDR Annex VIII 2.2
- MDR Annex VIII 2.3
- MDR Article 2 (5)
- MDR Annex VIII 2.6
- MDR Annex VIII 2.7
- MDR Article 2 (4)
- MDCG 2019-11
- MDR Annex VIII 2.4
- MDR Annex VIII 2.5
- MDR Annex VIII 3.7
- N.B. they are qualified as devices.
- Council Directive 80/181/EEC of 20 December 1979 on the approximation of the laws of the Member States relating to units of measurement and on the repeal of Directive 71/354/EEC (OJ L 39, 15.2.1980, p. 40).
- MDR article 2(10)
- MDR article 2(11)
- Rauscher et al., An overview of concepts and terms used in the European Commission's definition of nanomaterial, EUR 29647 EN, European Commission, JRC, Ispra, 2018,ISBN 978-92-79-99660-3, doi: 10.2760/459136, JRC 113469
- MDR Annex VIII 3.5
- Article 51 MDR
- 'Active therapeutic devices with an integrated or incorporated diagnostic function which significantly determines the patient management by the device, such as closed loop systems or automated external defibrillators, are classified as class III.'
- 2017/745/EC preamble, recital 15
- Coating containing nanomaterials